


Abstract
We predict the adsorption proficiency of hexagonal boron nitride (h-BN) sheets to toxic gas molecules like CO2, H2S and NO2 on the basis of first-principles density functional theory calculations. The computed energies predict the pristine h-BN sheet to have very little affinity towards the mentioned gas molecules. However, while doping C at the N site of the h-BN sheet brings a significant enhancement to the estimated adsorption energies, doping C at B site of the sheet is found to be energetically not so favorable. To have a higher coverage effect, the concentration of C doping on the h-BN sheet is further increased which resulted in upsurging the adsorption energies for the mentioned gas molecules. Among the three, CO2, H2S are found to be physisorbed to the C-doped h-BN sheets, where as the C-doped sheets are found to have strong affinity towards NO2 gas molecules.
Export citation and abstract BibTeX RIS
Introduction
Though graphene's unique electronic properties allow its applications in countless areas, the major challenge for creation of a tunable electronic band gap limits its use in various applications. In this respect, hexagonal boron-nitride (h-BN), having a very similar lattice to that of graphene, where the carbon atoms get swapped with covalently bonded alternate boron and nitrogen atoms, has a much larger band gap [1,2]. Thus, h-BN turns out to be a promising alternative which also has already been successfully synthesized by different experimental groups [1,3]. Like its carbon counterpart, the wide-band-gap (4.5–7 eV) [1,2] h-BN sheet also exhibits higher chemical inertness and thermal stability. It also exhibits some unique and fascinating properties like mechanical strength, thermal conductivity, chemical stability, and capability of withstanding higher temperature (upto  ) [4]. Though similar to graphene in many aspects, the h-BN sheet is insulating with a large direct band gap which is due to the strong nature of the B-N bonds. However, upon hydrogenation the band gap of h-BN sheets reduces rapidly which is quite opposite from what happens during graphene-to-graphane transition [5–8]. The electronic and mechanical properties of the h-BN can further be modulated by functionalizing the sheet with different atoms, molecules and various groups which makes the sheet an interesting material for various applications [1,2,9–12]. Owing to its stability, large surface area and great resistance to oxidation, the h-BN sheet is a good choice for gas sensing applications. The basic principle of the h-BN sensor is similar to any solid-state sensor which measures the change in electronic properties of the sheet upon adsorption of an incident gas molecule. Further, the variation in sheet's charge concentration depends on the type of the incident gas which can be either a donor or an acceptor [13].
) [4]. Though similar to graphene in many aspects, the h-BN sheet is insulating with a large direct band gap which is due to the strong nature of the B-N bonds. However, upon hydrogenation the band gap of h-BN sheets reduces rapidly which is quite opposite from what happens during graphene-to-graphane transition [5–8]. The electronic and mechanical properties of the h-BN can further be modulated by functionalizing the sheet with different atoms, molecules and various groups which makes the sheet an interesting material for various applications [1,2,9–12]. Owing to its stability, large surface area and great resistance to oxidation, the h-BN sheet is a good choice for gas sensing applications. The basic principle of the h-BN sensor is similar to any solid-state sensor which measures the change in electronic properties of the sheet upon adsorption of an incident gas molecule. Further, the variation in sheet's charge concentration depends on the type of the incident gas which can be either a donor or an acceptor [13].
Using first-principles calculations, Bhattacharya et al. have discussed the band-gap engineering of the h-BN sheet by functionalizing the sheet with various functional groups like H, F, OH, CH3, CN and NH2. Functionalizing the h-BN sheet with CHO group also yields an appropriate band gap making the sheet suitable for many electronic device applications [14]. In another context, Deng et al. have studied the adsorption properties of various gas molecules on pure as well as carbon-doped g-BN sheet. With C doping, the binding strength of some of the selected gas molecules changes from the physisorption to the chemisorption range [15]. Depending upon the coverage effect, with hydrogen and fluorine adatoms the band gap of the BN sheet gets reduced from 4.7 eV to a low value of 0.6 eV [16]. In another context, Tang et al. [17] have reported a novel technique to tune the band gap of the BN nano-structure upon adsorption of organic molecules. One of the most convenient way to modify the adsorption properties of the h-BN sheet is to dope the sheet with carbon atoms. Several experiments have also reported successful synthesis of such C-doped BN nano-structures [18,19]. However, the gas sensing propensity of the h-BN sheet when doped with C atoms is so far the least discussed in the literature.
Among many, H2S and NO2 are considered as highly toxic gases having adverse effects on humans lives. Environments with H2S concentration higher than 250 ppm [20] and that of NO2 higher than even 1 ppm [20] are found to be quite fatal. Moreover, CO2 is one of the main causes of greenhouse effect, and mostly results from burning of fossil fuels. Thus, designing a sensor capable of detecting the presence of H2S, NO2 and CO2 even in small quantity is highly desirable. In this work, using the density functional theory (DFT), we have calculated the adsorption mechanism of CO2, H2S and NO2 gas molecules on pure as well as carbon-doped h-BN sheets. The absorption energies of the above-mentioned gases with respect to the pristine h-BN sheet are found to be very small which affirms the sheet to be unsuitable for the gas sensing purpose. In the next step, we have doped the sheet with C atom and while doping, the N site is found to be more favorable than that of the B site of the h-BN sheet. With C-doped sheets, we find the absorption energies of the mentioned gas molecules to be suitable enough for sensing them and found to be the most suitable for the NO2 gas molecules.
Computational methods
For this work, we have modeled a  boron nitride super cell consisting of 32 atoms with a vacuum space of 15 Å along the conventional (001) direction to avoid the unwanted interaction between the periodically repeating units (fig. 1(a)). To get C-doped h-BN sheets, we have substituted C at the N site of the BN sheet. We have performed spin-polarized DFT calculation by using the Vienna ab initio simulation package (VASP) [21–23]. The core-valence interactions are treated with the projector augmented-wave (PAW) method [24] and the exchange-correlation part is approximated with the generalized gradient approximation (GGA) of Perder-Wang-Ernzerhof (PBE) [25]. For the valence electron configurations the pseudopotentials were chosen to be
boron nitride super cell consisting of 32 atoms with a vacuum space of 15 Å along the conventional (001) direction to avoid the unwanted interaction between the periodically repeating units (fig. 1(a)). To get C-doped h-BN sheets, we have substituted C at the N site of the BN sheet. We have performed spin-polarized DFT calculation by using the Vienna ab initio simulation package (VASP) [21–23]. The core-valence interactions are treated with the projector augmented-wave (PAW) method [24] and the exchange-correlation part is approximated with the generalized gradient approximation (GGA) of Perder-Wang-Ernzerhof (PBE) [25]. For the valence electron configurations the pseudopotentials were chosen to be  ,
,  ,
,  ,
,  , 2s1, and
, 2s1, and  for B, N, C, O, H, and S elements, respectively. During the structure optimization, the
for B, N, C, O, H, and S elements, respectively. During the structure optimization, the  k-points mesh is used to sample the entire Brillouin zone within the Monkhorst-Pack (MP) scheme [26]. However, for an accurate electronic density of states (DOS), the energy optimization is done with a denser k-points mesh of
k-points mesh is used to sample the entire Brillouin zone within the Monkhorst-Pack (MP) scheme [26]. However, for an accurate electronic density of states (DOS), the energy optimization is done with a denser k-points mesh of  within the MP scheme. For achieving the ground-state configurations for the pristine as well as C-doped h-BN sheets, the criterion for the Hellmann-Feynman force on each atom in the supercell was taken to be less than 0.01 eV/Å, and the self-consistence energy convergence is taken to be
within the MP scheme. For achieving the ground-state configurations for the pristine as well as C-doped h-BN sheets, the criterion for the Hellmann-Feynman force on each atom in the supercell was taken to be less than 0.01 eV/Å, and the self-consistence energy convergence is taken to be  . The symmetry constraints were switched off and an energy cut-off of 600 eV was used for the plane-wave basis set.
. The symmetry constraints were switched off and an energy cut-off of 600 eV was used for the plane-wave basis set.
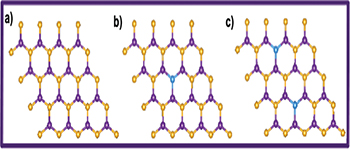
Fig. 1: (Color online) Top view of pristine h-BN, CBN and 2CBN sheets, respectively. Violet: B; gold: N; blue: C.
Download figure:
Standard imageIn the current study, we aim at calculating the adsorption energies of CO2, H2S and NO2 gas molecules on pristine as well as on C-doped h-BN sheets. For such weakly interacting systems, the GGA approximation usually ends up with underestimating the adsorption energies, whereas the local density approximation (LDA) rather overestimates the binding energies. While dealing with such limitations within the DFT but to have a better estimation for the adsorption energies, we have incorporated the van der Waals type corrections, as implemented within the DFT-D2 approach, to our calculations [27]. The DFT-D2 approach adds a semi-empirical dispersion term to the conventional Kohn-Sham energy. The adsorption energies of the mentioned molecules to the pristine and C-doped h-BN sheets are estimated as
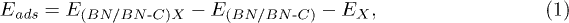
where,  is the total DFT energy of pristine/C-doped h-BN sheets while adsorbed with incident gas molecules.
is the total DFT energy of pristine/C-doped h-BN sheets while adsorbed with incident gas molecules.  is the total DFT energy of pristine/C-doped h-BN bare sheets and EX represents the DFT energy of the incident gas molecule, where X can be CO2, H2S or NO2.
is the total DFT energy of pristine/C-doped h-BN bare sheets and EX represents the DFT energy of the incident gas molecule, where X can be CO2, H2S or NO2.
Results and discussion
The optimized structures of pristine h-BN (fig. 1(a)) and that of C-doped h-BN sheets (fig. 1(b), (c)) are depicted in fig. 1. While doping C to the h-BN sheet, in the first step we have replaced one N atom with a C atom and in the next step two N atoms with two C atoms to get stable CBN and 2CBN sheets, respectively. Successful synthesis of such C-doped BN structures has already been reported elsewhere [28]. However, the C doping at the B sites of the h-BN sheets are found to be energetically rather unsuitable. The formation energy at the N substitution site (0.89 eV) is found to be much lower than that at the B substitution (2.29 eV) site [29]. In the case of the h-BN sheet, the B-N bond length is estimated to be 1.44 Å. Whereas, in the optimized structures of CBN and 2CBN sheets, the B-C bond lengths are calculated to be 1.49 Å and 1.48 Å, respectively. In the case of 2CBN, the C-C bond length is found to be 4.98 Å which is large enough to have a uniform distribution of C atoms in the h-BN sheet. We have the spin-polarized partial density of states (PDOS) of h-BN, CBN and 2CBN sheets as shown in fig. 2(a)–(c). In the case of h-BN, the valence band region is mostly dominated by N(2p) states whereas in CBN, the valence band region is mostly dominated by B(2p)-C(2p) hybrid bands (fig. 2(b)). The impurity states due to C(2p) are available in the conduction band region. On the other hand, in 2CBN, both valence band and conduction band regions are mostly dominated by impurity C(2p) states. The C(2p) states get hybridized with N(2p) states as shown in fig. 2(c). The calculated adsorption energies of H2S, CO2 and NO2 molecules (with different coverage effect) when exposed to h-BN as well as C-doped BN sheets are summarized in table 1. Here onwards, we have discussed the affinity of h-BN, CBN and 2CBN sheets towards H2S, CO2 and NO2 toxic gas molecules, separately. We have calculated the adsorption energy by using eq. (1); and to estimate the charge transfer between the gas molecules and the sheets we have used the Bader charge analysis [30].

Fig. 2: (Color online) PDOS of h-BN (a), CBN (b) and 2CBN (c) sheets, respectively.
Download figure:
Standard imageTable 1:.
The adsorption energies  in eV calculated with GGA and van der Waals (Vdw) approaches. h-BN, CBN and 2CBN are the pristine, one-C-doped and two-C-doped BN sheets, respectively.
in eV calculated with GGA and van der Waals (Vdw) approaches. h-BN, CBN and 2CBN are the pristine, one-C-doped and two-C-doped BN sheets, respectively.
| Sheets | H2S adsorbed (eV/mol) | CO2 adsorbed (eV/mol) | NO2 adsorbed (eV/mol) |
|---|---|---|---|
| BN | −0.11 | −0.20 | −0.55 |
| CBN | −0.70 | − | −2.9 |
| 2CBN | −0.55 | −0.28 | −2.4 |
H2S
Here, the most preferential binding site of H2S on the h-BN sheet was obtained by considering different positions like B-top, N-top, bridge-centre (centre of the B-N bond) and hollow-centre (centre of the hexagon formed by B and N atoms) sites. The optimum orientation of H2S on the h-BN sheet was obtained by considering the energetics while pointing S and then the H atom towards the sheet. It is found that the H2S prefers to sit on the hollow-centre site of the h-BN sheet, with the S atom pointing towards the sheet as shown in fig. 3. The optimum H2S-to-sheet distance is found to be 3.12 Å. The H2S undergoes a slight structural distortion with the S-H bond length changing from 1.34 Å to 1.35 Å; and the H-S-H bond angle decreases from  to
to  . The adsorption energy of H2S to the h-BN sheet (BN-H2S) with van der Waals corrections is calculated to be
. The adsorption energy of H2S to the h-BN sheet (BN-H2S) with van der Waals corrections is calculated to be  . The negative sign indicates an exothermic behavior for the H2S adsorption. Further, we calculate the adsorption energies of H2S on CBN (CBN-H2S) and 2CBN (2CBN-H2S) sheets. For the CBN-H2S sheet, the adsorption increases to
. The negative sign indicates an exothermic behavior for the H2S adsorption. Further, we calculate the adsorption energies of H2S on CBN (CBN-H2S) and 2CBN (2CBN-H2S) sheets. For the CBN-H2S sheet, the adsorption increases to  whereas the optimized distance of CBN to H2S is estimated to be 3.02 Å. In the case of 2CBN-2H2S sheet, the two H2S molecules get adsorbed at the top of two doped C atoms and the adsorption energy per molecule is estimated to be
whereas the optimized distance of CBN to H2S is estimated to be 3.02 Å. In the case of 2CBN-2H2S sheet, the two H2S molecules get adsorbed at the top of two doped C atoms and the adsorption energy per molecule is estimated to be  . From the PDOS plots (fig. 4(a)) of BN-H2S, one can see a small hybridization between N(p) and S(p). Whereas, in CBN-H2S, there is a stronger hybridization between C(p), N(p) of the sheet with
. From the PDOS plots (fig. 4(a)) of BN-H2S, one can see a small hybridization between N(p) and S(p). Whereas, in CBN-H2S, there is a stronger hybridization between C(p), N(p) of the sheet with  resulting in a higher adsorption energy (
resulting in a higher adsorption energy ( , table 1). In 2CBN-H2S, there are more C(p) states near the Fermi energy. The doped C atoms are rather more tightly bound to both the B and N atoms of the sheet in comparison with the H2S molecule. Thus, the adsorption energy in 2CBN-H2S rather decreases (
, table 1). In 2CBN-H2S, there are more C(p) states near the Fermi energy. The doped C atoms are rather more tightly bound to both the B and N atoms of the sheet in comparison with the H2S molecule. Thus, the adsorption energy in 2CBN-H2S rather decreases ( , table 1) compared to that in CBN-2H2S. Further, from Bader charge analysis we found that in BN-H2S a small amount of charge is transferred from the H2S molecule to the sheet. In CBN-H2S, there is a charge transfer of 0.07e, whereas for 2CBN-H2S almost no charge transfer occurs from the H2S molecules to the sheets.
, table 1) compared to that in CBN-2H2S. Further, from Bader charge analysis we found that in BN-H2S a small amount of charge is transferred from the H2S molecule to the sheet. In CBN-H2S, there is a charge transfer of 0.07e, whereas for 2CBN-H2S almost no charge transfer occurs from the H2S molecules to the sheets.
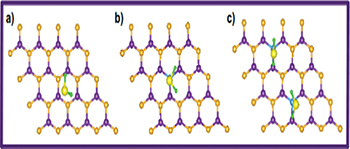
Fig. 3: (Color online) Top view of BN-H2S, CBN-H2S and 2CBN-2H2S sheets, respectively. Violet: B; gold: N; blue: C; yellow: S; green: H.
Download figure:
Standard image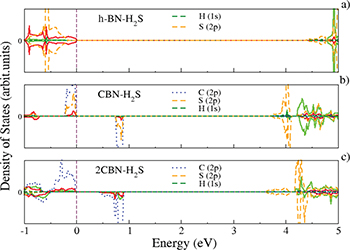
Fig. 4: (Color online) PDOS plots of BN-H2S (a), CBN-H2S (b) and 2CBN-2H2S (c) sheets, respectively.
Download figure:
Standard imageCO2
Similar to H2S, we have also found the most preferential site of CO2 on the h-BN sheet (BN-CO2). The energetics reveal the hollow site, with a binding energy of  , and a BN-to-CO2 distance of 3.1 Å as the most suitable configuration for the BN-CO2 system. In BN-CO2, CO2 undergoes a structural relaxation where the C-O bond length changes from 1.16 Å to 1.20 Å. The adsorption energies of CBN-CO2 and 2CBN-2CO2 are summarized in table 1. Unlike H2S, the adsorption energies of CO2 do not improve with C doping. In 2CBN-2CO2, the adsorption energy per molecule is calculated to be
, and a BN-to-CO2 distance of 3.1 Å as the most suitable configuration for the BN-CO2 system. In BN-CO2, CO2 undergoes a structural relaxation where the C-O bond length changes from 1.16 Å to 1.20 Å. The adsorption energies of CBN-CO2 and 2CBN-2CO2 are summarized in table 1. Unlike H2S, the adsorption energies of CO2 do not improve with C doping. In 2CBN-2CO2, the adsorption energy per molecule is calculated to be  . The small value of adsorption energy indicates the physisorption of CO2 to h-BN and C-doped h-BN sheets. Contrary to H2S absorbed sheets, the charge transfer occurs rather from the sheets towards the respective CO2 molecule (see fig. 5). To be precise, 0.01e, 0.04e and 0.02e are the charge being transferred from h-BN, CBN and 2CBN sheets to the respective CO2 molecules. We have the PDOS plots of BN-CO2, CBN-CO2 and 2CBN-2CO2 as shown in fig. 6(a)–(c). Compared to BN-CO2 (panel (a)), in CBN-CO2 and 2CBN-2CO2 there are enough available states for C near the Fermi energy. We see a coupling between the C states and the N states of the sheet which leads to physisorption of CO2 to the C-doped h-BN sheet.
. The small value of adsorption energy indicates the physisorption of CO2 to h-BN and C-doped h-BN sheets. Contrary to H2S absorbed sheets, the charge transfer occurs rather from the sheets towards the respective CO2 molecule (see fig. 5). To be precise, 0.01e, 0.04e and 0.02e are the charge being transferred from h-BN, CBN and 2CBN sheets to the respective CO2 molecules. We have the PDOS plots of BN-CO2, CBN-CO2 and 2CBN-2CO2 as shown in fig. 6(a)–(c). Compared to BN-CO2 (panel (a)), in CBN-CO2 and 2CBN-2CO2 there are enough available states for C near the Fermi energy. We see a coupling between the C states and the N states of the sheet which leads to physisorption of CO2 to the C-doped h-BN sheet.

Fig. 5: (Color online) Top view of BN-CO2 (a), CBN-CO2 (b) and 2CBN-2CO2 (c) sheets, respectively. Violet: B; gold: N; blue: C; red: O.
Download figure:
Standard image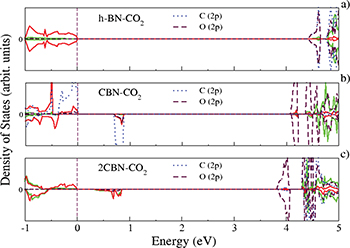
Fig. 6: (Color online) PDOS plots of BN-CO2 (a), CBN-CO2 (b) and 2CBN-2CO2 (c) sheets, respectively.
Download figure:
Standard imageNO2
Similar to CO2 and H2S adsorption, we have also calculated the adsorption energies of NO2 on h-BN, CBN and 2CBN sheets in different configurations. Here, the hollow site, with the N atom of NO2 pointing towards the sheet is found to be the most stable configuration with an adsorption energy of  and a BN-NO2 distance of 3 Å. The NO2 molecule undergoes a structural change with the N-O bond distance changing from 1.23 Å to 1.26 Å; and the O-N-O angle changes from
and a BN-NO2 distance of 3 Å. The NO2 molecule undergoes a structural change with the N-O bond distance changing from 1.23 Å to 1.26 Å; and the O-N-O angle changes from  to
to  . However, with C doping, there is a drastic improvement in the adsorption energies of NO2 in CBN-NO2 and 2CBN-NO2 sheets. For CBN-NO2 and 2CBN-2NO2, the adsorption energies are calculated to be
. However, with C doping, there is a drastic improvement in the adsorption energies of NO2 in CBN-NO2 and 2CBN-NO2 sheets. For CBN-NO2 and 2CBN-2NO2, the adsorption energies are calculated to be  and
and  , respectively (table 1), indicating the chemisorption of NO2 to C-doped BN sheets. The corresponding NO2-to-CBN and -2CBN distances are found to be 1.54 Å and 1.52 Å, respectively (see fig. 7). The PDOS plots of BN-NO2, CBN-NO2 and 2CBN-2NO2 are shown in fig. 8(a), (b), (c), respectively. In BN-NO2, the available valence states near the Fermi level are N(p) states. Whereas in CBN-NO2 and 2CBN-2NO2 the valence band region is mostly dominated by C(p), N(p) as well as O(p) states, indicating a strong hybridization between the states which strongly binds NO2 to the sheets. Like CO2, NO2 gathers electric charge from the respective h-BN and C-doped BN sheets. In case of BN-NO2, a charge of 0.40e gets transferred from the sheet to the NO2 molecule. Whereas in CBN-NO2 and 2CBN-2NO2 a significantly large amount of charge equal to 0.55e and 0.6e gets transferred to the respective NO2 molecules.
, respectively (table 1), indicating the chemisorption of NO2 to C-doped BN sheets. The corresponding NO2-to-CBN and -2CBN distances are found to be 1.54 Å and 1.52 Å, respectively (see fig. 7). The PDOS plots of BN-NO2, CBN-NO2 and 2CBN-2NO2 are shown in fig. 8(a), (b), (c), respectively. In BN-NO2, the available valence states near the Fermi level are N(p) states. Whereas in CBN-NO2 and 2CBN-2NO2 the valence band region is mostly dominated by C(p), N(p) as well as O(p) states, indicating a strong hybridization between the states which strongly binds NO2 to the sheets. Like CO2, NO2 gathers electric charge from the respective h-BN and C-doped BN sheets. In case of BN-NO2, a charge of 0.40e gets transferred from the sheet to the NO2 molecule. Whereas in CBN-NO2 and 2CBN-2NO2 a significantly large amount of charge equal to 0.55e and 0.6e gets transferred to the respective NO2 molecules.
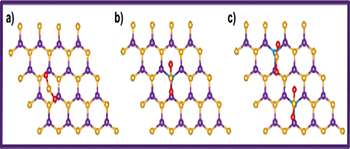
Fig. 7: (Color online) Top view of BN-NO2 (a), CBN-NO2 (b) and 2CBN-2NO2 (c) sheets, respectively. Violet: B; gold: N; blue: C; red: O.
Download figure:
Standard image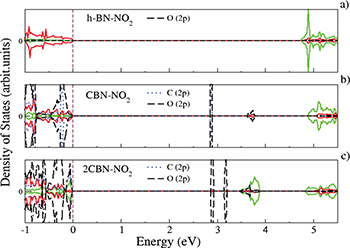
Fig. 8: (Color online) PDOS plots of BN-NO2 (a), CBN-NO2 (b) and 2CBN-2NO2 (c) sheets, respectively.
Download figure:
Standard imageSummary
In summary, we find that the pristine h-BN sheet has less affinity towards H2S, CO2 and NO2 gas molecules. However, the adsorption energies get improved significantly when doping the h-BN sheet with C atoms. The C doping at the N site of the h-BN sheet is found to be energetically preferred over the doping at the B site. For one C-doped (CBN) and two C-doped (2CBN) sheets, the H2S, and CO2 gas molecules get physisorbed whereas NO2 gets chemisorbed. In the case of H2S, the doping with a single C atom enhances the C(p), N(p) and S(p) hybridization: the adsorption energy increases. Whereas doping with two C atoms, the C(p)-N(p) hybridization gets stronger while C(p)-S(p) bonding gets weaker: the adsorption energy decreases. For NO2, there is a strong hybridization among C(p), N(p) and O(p) states with a significant charge transfer occurring from the sheet to NO2; thus resulting in chemisorption. However, with C doping the adsorption energies of CO2 to h-BN sheets do not change significantly.
Acknowledgments
We are grateful to STINT for financial support. MS is thankful to MARCO XXI-Erasmus Mundus in Central Asia for a PhD scholarship and PP would like to thank the Swedish Institute for a postdoctoral fellowship. We would like to acknowledge the Carl Tryggers Stiftelse for Vetenskaplig Forskning (CTS), the Swedish Research Council (VR) and the Swedish Energy Agency for financial support. We are also thankful to NSC, HPC2N and UPPMAX for providing us with computing resources.