


Abstract
We study the electromigration-induced drift of monolayer Ag islands on Ag(111) which contain one Cu atom. For this purpose a three-dimensional self-learning kinetic Monte Carlo model was extended, and a realistic many-body potential was used. The only free parameters of the model are the effective valences of the Ag and Cu atoms. Due to the impurity, the island drift is significantly reduced, especially for small islands. This is traced back to sequential pinning and depinning events, which are analyzed in detail. Surprisingly, this phenomenon is qualitatively independent of the impurity's effective valence, as long as the impurity does not detach from the island edge. How strongly the drift velocity is reduced depends on the effective valence.
Export citation and abstract BibTeX RIS
Introduction
High current densities in metal conductors can lead to a drift of the metal ions. This effect is known as electromigration (EM). EM is a major reliability issue for interconnects in integrated circuits [1–3] but can also be used to manipulate nanostructures in a desired way [4–6]. Empirically, the reason for the ion drift is an electromigration force  , which is proportional to the local electric field
, which is proportional to the local electric field  [7]:
[7]:

In this equation,  denotes the effective valence of the metal ions and e is the elementary charge. The sign of
denotes the effective valence of the metal ions and e is the elementary charge. The sign of  characterizes the direction of the ion drift. For many metals, like silver (Ag), one finds theoretically and experimentally
characterizes the direction of the ion drift. For many metals, like silver (Ag), one finds theoretically and experimentally  [7–10]. This implies a drift opposite to the direction of
[7–10]. This implies a drift opposite to the direction of  .
.
It has long been known empirically that impurity atoms can reduce electromigration [1,2,11] and therefore increase the lifetime of interconnects. However, EM simulations with more than one atomic species represent a computational challenge, which so far has rarely been addressed, and if so, only with simplified models. For example, in [10,12–16] the electromigration of clusters (islands or voids) is considered only for atoms of one type. Electromigration of an impurity atom in the bulk has been studied with the so-called five-frequency model [17]. By means of ab initio calculations, Bevan et al. [18] showed that certain atomic processes have reduced rates, when impurities are involved. The present paper presents the first material-specific simulation of electromigration of islands on a surface in the presence of an impurity. This is achieved by extending the self-learning kinetic Monte Carlo (SLKMC) method [19–21] for copper (Cu)-contaminated Ag islands on Ag(111).
Simulation model and setup
We generalize our 3D SLKMC model such that the temporal evolution of solids with two kinds of atoms (A and B) can be simulated. In our specific study, atom type A corresponds to Ag and atom type B corresponds to Cu.
In the SLKMC model thermally activated atom hops are performed with a rate ν given by an Arrhenius law:

In this equation  is the Boltzmann constant, and T is the absolute temperature. The attempt frequency is assumed to be independent of the atomic process,
is the Boltzmann constant, and T is the absolute temperature. The attempt frequency is assumed to be independent of the atomic process,  [22], and the activation energies
[22], and the activation energies  will be calculated from a suitable potential. The attempt frequency and the activation energy have errors. However, as long as the effect on the ratio between any two rates is weak, the error amounts essentially to a rescaling of time. The SLKMC model differs from commonly used KMC implementations [22,23] by combining the accuracy of rates calculated from a realistic potential with the efficiency of a rate catalog. The activation energies are calculated by means of the drag method [24] and saved in a database, from which they are retrieved, when needed during the simulation. In case, a configuration is encountered, which cannot be found in the database yet, the corresponding activation energies are calculated on the fly and added to the rate catalog. To ensure fast addressing within the rate catalog, bit-coding of the hopping processes is used. They are characterized by the occupied lattice sites of two shells surrounding the initial and final position of the hopping atom.
will be calculated from a suitable potential. The attempt frequency and the activation energy have errors. However, as long as the effect on the ratio between any two rates is weak, the error amounts essentially to a rescaling of time. The SLKMC model differs from commonly used KMC implementations [22,23] by combining the accuracy of rates calculated from a realistic potential with the efficiency of a rate catalog. The activation energies are calculated by means of the drag method [24] and saved in a database, from which they are retrieved, when needed during the simulation. In case, a configuration is encountered, which cannot be found in the database yet, the corresponding activation energies are calculated on the fly and added to the rate catalog. To ensure fast addressing within the rate catalog, bit-coding of the hopping processes is used. They are characterized by the occupied lattice sites of two shells surrounding the initial and final position of the hopping atom.
To identify a hopping process within an atom configuration of types A and B, we use five indices: the first one corresponds to the hopping direction, the second and third correspond to the two shells occupied with atoms of type A and the fourth and fifth correspond to the two shells occupied with atoms of type B. An example of this coding is shown in fig. 1. In this example, one atom of type B is embedded within atoms of type A. However, this coding is general enough to consider more than one atom of type B.
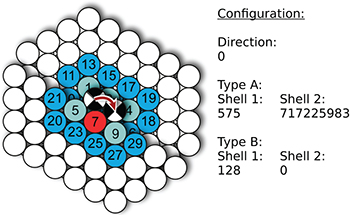
Fig. 1: (Color online) Example of a bit-coded two-type atom configuration. Atoms of type A are shown in blue. The light blue circles represent occupied lattice sites of the first shell, the dark blue circles represent occupied lattice sites of the second shell. The red circle represents a site of the first shell occupied by an atom of type B. White circles represent atoms, which remain stationary during the calculation of  .
.
Download figure:
Standard imageWe implemented an EAM potential [25,26] in order to capture the many-body nature of metallic bonding correctly. We used a well-tested version of it for Ag-Cu-alloys by Williams et al. [27].
In order to simulate EM, we modify the energy term in eq. (2) by  times the vector connecting the initial hopping position and the saddle point
times the vector connecting the initial hopping position and the saddle point  :
:

The values of  are also gathered during the calculation of
are also gathered during the calculation of  .
.
The simulations are performed at  . Three parameters are varied. First, the number N of embedded Cu atoms (
. Three parameters are varied. First, the number N of embedded Cu atoms ( or 1), and second, the radius R of the island (
or 1), and second, the radius R of the island ( to 15, where
to 15, where  is the nearest-neighbor distance of Ag at room temperature [28]). The third parameter is the ratio of effective valences
is the nearest-neighbor distance of Ag at room temperature [28]). The third parameter is the ratio of effective valences  . For the physical system considered it has a fixed value, which, however, is not known. Therefore, we consider values in the range
. For the physical system considered it has a fixed value, which, however, is not known. Therefore, we consider values in the range  . Thus, we can also assess qualitatively how the sign and relative strength of the electromigration forces influences the island drift. We also consider negative ratios, because for Ag mass transport in the direction of
. Thus, we can also assess qualitatively how the sign and relative strength of the electromigration forces influences the island drift. We also consider negative ratios, because for Ag mass transport in the direction of  or
or  was observed [10,29]. The broad range of ratios reflects the large deviations of experimentally known
was observed [10,29]. The broad range of ratios reflects the large deviations of experimentally known  [9]. For monolayer islands on a flat metallic surface it is legitimate to assume a constant electric field [14]. Therefore,
[9]. For monolayer islands on a flat metallic surface it is legitimate to assume a constant electric field [14]. Therefore,  . For Ag atoms we set
. For Ag atoms we set  as in [16].
as in [16].
For the investigated systems, the dominant mode of mass transport is edge diffusion. Although considered in the model, an additional terrace diffusion can be neglected [16].
In our simulations we are interested in the drift velocity parallel to  . As we set
. As we set  (where
(where  is the x-unit vector) we calculate the x-component of the drift velocity vx. To do so, the temporal evolution of the x-center-of-mass coordinate rx of the islands is investigated.
is the x-unit vector) we calculate the x-component of the drift velocity vx. To do so, the temporal evolution of the x-center-of-mass coordinate rx of the islands is investigated.
Every simulation of each set of parameters is repeated ten times. Subsequently, the ten  trajectories are averaged to calculate the mean velocity
trajectories are averaged to calculate the mean velocity  .
.
Results
The mean drift velocities and the corresponding error bars are shown in fig. 2 for each set of parameters. The results of pure Ag islands are in accordance to the ones found in [16]. For larger islands  we expect
we expect  [16]. If an Ag island contains one Cu atom, our simulations reveal that
[16]. If an Ag island contains one Cu atom, our simulations reveal that  decreases. The ratio
decreases. The ratio  depends on R and becomes larger with decreasing R: At
depends on R and becomes larger with decreasing R: At  and
and  a maximal ratio of approximately 40 is observed. The drift reduction is only weakly influenced by
a maximal ratio of approximately 40 is observed. The drift reduction is only weakly influenced by  : qualitatively, higher values lead to higher drift velocities. The results for
: qualitatively, higher values lead to higher drift velocities. The results for  seem to be outliers. But as Heinonen et al. found in [30], the diffusion constant D shows size-dependent oscillations for small islands. Since
seem to be outliers. But as Heinonen et al. found in [30], the diffusion constant D shows size-dependent oscillations for small islands. Since  , we see such an oscillation.
, we see such an oscillation.
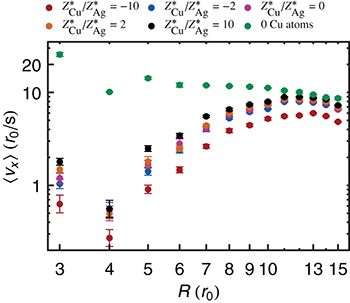
Fig. 2: (Color online) All calculated  values on a log-log scale. Some error bars are of the size of the plot symbols. The results indicate that the EM-induced drift velocity of islands including one Cu atom are reduced for a considerably amount compared to pure Ag islands.
values on a log-log scale. Some error bars are of the size of the plot symbols. The results indicate that the EM-induced drift velocity of islands including one Cu atom are reduced for a considerably amount compared to pure Ag islands.
Download figure:
Standard imageThe reason for the drift reduction induced by a Cu impurity can be understood by investigating single  trajectories. Two representative trajectories are shown in fig. 3 (
trajectories. Two representative trajectories are shown in fig. 3 ( ,
,  or −10). In addition, the trajectory of a pure Ag island without a Cu impurity is plotted.
or −10). In addition, the trajectory of a pure Ag island without a Cu impurity is plotted.

Fig. 3: (Color online)  trajectories for islands with
trajectories for islands with  . The black and red trajectories show the motion of Ag-Cu islands with the ratios
. The black and red trajectories show the motion of Ag-Cu islands with the ratios  and −10, respectively. The green line corresponds to a trajectory of a pure Ag island. Due to a pinning, the trajectories of both Ag-Cu islands show plateaus. After a depinning, the velocities of the Ag-Cu islands match the one of the pure Ag island.
and −10, respectively. The green line corresponds to a trajectory of a pure Ag island. Due to a pinning, the trajectories of both Ag-Cu islands show plateaus. After a depinning, the velocities of the Ag-Cu islands match the one of the pure Ag island.
Download figure:
Standard imageIn contrast to the pure Ag island, the trajectories of both Ag-Cu islands show plateaus. This is due to a pinning of the whole island.
The pinning lasts for a considerable but finite time (plateaus). After this pinning time τ, a depinning occurs. Once the depinning happened, both Ag-Cu islands migrate with a mean drift velocity  which is within the standard error of the mean drift velocity of pure Ag islands
which is within the standard error of the mean drift velocity of pure Ag islands  . The pinning time τ and migration range between two plateaus
. The pinning time τ and migration range between two plateaus  depend on
depend on  . The plotted trajectories show an increase of τ and a decrease of
. The plotted trajectories show an increase of τ and a decrease of  with
with  , respectively. Therefore, we see a dependence of
, respectively. Therefore, we see a dependence of  on the ratios as is shown in fig. 2.
on the ratios as is shown in fig. 2.
To understand the pinning effect in more detail, we discuss five representative simulation snapshots of an Ag-Cu island with  and their qualitative temporal occurrence (see fig. 4).
and their qualitative temporal occurrence (see fig. 4).

Fig. 4: (Color online) Five representative simulation snapshots and their qualitatively temporal occurrence. An Ag-Cu island (blue and red) with  is placed on a Ag(111) surface (gray). (a) Start configuration with a Cu atom placed at an arbitrary site within the island. (b) Rightward migration of Ag atoms until the Cu atom becomes the second nearest neighbor of a left-edge atom. The pinning takes place. (c) Due to random fluctuations the Cu atom itself becomes an edge atom. (d) The Cu atom diffuses along the island edge. (e) The Ag atoms migrate again.
is placed on a Ag(111) surface (gray). (a) Start configuration with a Cu atom placed at an arbitrary site within the island. (b) Rightward migration of Ag atoms until the Cu atom becomes the second nearest neighbor of a left-edge atom. The pinning takes place. (c) Due to random fluctuations the Cu atom itself becomes an edge atom. (d) The Cu atom diffuses along the island edge. (e) The Ag atoms migrate again.
Download figure:
Standard imageA simulation starts at a time t0 with a round island as shown in fig. 4(a). The Cu atom is placed at an arbitrary site within the island.
 leads to a drift of Ag atoms to the right via edge diffusion. Therefore, the whole island migrates rightwards until the Cu atom becomes the second nearest neighbor of a left-edge atom. This case is shown in fig. 4(b) and occurs at a time t1. The difference between t0 and t1 is rather short and is determined by
leads to a drift of Ag atoms to the right via edge diffusion. Therefore, the whole island migrates rightwards until the Cu atom becomes the second nearest neighbor of a left-edge atom. This case is shown in fig. 4(b) and occurs at a time t1. The difference between t0 and t1 is rather short and is determined by  . As mentioned before, our SLKMC model considers two shells surrounding a hopping atom. Therefore, the Cu atom does not influence the edge diffusion until t1. After t1, the Cu atom influences the hopping processes of its surrounding Ag atoms. It turns out that many calculated activation energies for configurations including a Cu atom are higher than for a pure Ag configuration (note that for different impurities this may not be the case [18]). Therefore, the pinning starts at t1.
. As mentioned before, our SLKMC model considers two shells surrounding a hopping atom. Therefore, the Cu atom does not influence the edge diffusion until t1. After t1, the Cu atom influences the hopping processes of its surrounding Ag atoms. It turns out that many calculated activation energies for configurations including a Cu atom are higher than for a pure Ag configuration (note that for different impurities this may not be the case [18]). Therefore, the pinning starts at t1.
Due to random fluctuations the Cu atom can become an edge atom. This case is shown in fig. 4(c) and occurs at a time t2. Then the Cu atom can diffuse along the island edge which leads to depinning. Whether and how far it diffuses, depends on the ratio  . In our simulations, the Cu atom is strongly bound to the island, so that even for
. In our simulations, the Cu atom is strongly bound to the island, so that even for  we never observed that it detaches from the island edge. Thus, a variation of
we never observed that it detaches from the island edge. Thus, a variation of  only affects the distance, the Cu atom diffuses, before it becomes surrounded again by Ag atoms. The larger
only affects the distance, the Cu atom diffuses, before it becomes surrounded again by Ag atoms. The larger  , the more likely a Cu edge atom will hop in the direction of
, the more likely a Cu edge atom will hop in the direction of  . For
. For  it is very likely that the Cu atom reaches the opposite island edge. Thus, we see
it is very likely that the Cu atom reaches the opposite island edge. Thus, we see  for
for  in fig. 3. In the case of
in fig. 3. In the case of  the migration range
the migration range  becomes small or even zero. We see
becomes small or even zero. We see  if the Cu atom becomes surrounded again, before it diffuses in the direction of
if the Cu atom becomes surrounded again, before it diffuses in the direction of  .
.
In fig. 4(d) the Cu atom had diffused about  along the island edge. The Cu diffusion ends at a time t3. The difference
along the island edge. The Cu diffusion ends at a time t3. The difference  is small compared to
is small compared to  because edge diffusion occurs at short time scales. The pinning time is characterized by
because edge diffusion occurs at short time scales. The pinning time is characterized by  .
.
After t3, when the Cu atom becomes embedded into the island again, the Ag atoms migrate freely with  . The snapshot in fig. 4(e) shows the Ag diffusion after the depinning at time t4. The pure Ag diffusion will last until the Cu atom becomes the second nearest neighbor of a left-edge atom. If this happens, a new pinning occurs and the described procedure takes place again.
. The snapshot in fig. 4(e) shows the Ag diffusion after the depinning at time t4. The pure Ag diffusion will last until the Cu atom becomes the second nearest neighbor of a left-edge atom. If this happens, a new pinning occurs and the described procedure takes place again.
We have discussed the case  . For other choices of R, we can also see a pinning and depinning of the island. But with increasing R it becomes more difficult to separate between the pinning and pure Ag drift since
. For other choices of R, we can also see a pinning and depinning of the island. But with increasing R it becomes more difficult to separate between the pinning and pure Ag drift since  decreases [16]. The rx trajectories approximate the ones of pure Ag islands and the difference between
decreases [16]. The rx trajectories approximate the ones of pure Ag islands and the difference between  and
and  becomes smaller as is shown in fig. 2.
becomes smaller as is shown in fig. 2.
Conclusion and outlook
Using an extended SLKMC model, we were able to simulate the electromigration-induced drift of monolayer Ag islands on Ag(111) which contain one Cu atom. Our simulations show that the single Cu atom causes a pinning of the whole island. The pinning occurs regardless of the value and sign of  and induces a reduction of the island drift velocity. The difference between
and induces a reduction of the island drift velocity. The difference between  and
and  is very large for small R but decreases, if R grows. This effect is also expected for different alloys and island structures. The pinning described in this paper depends crucially on the increase of activation energies due to an impurity nearby, such as already calculated by Bevan et al. [18] in a different context. For which alloys the effect will occur can be predicted by density functional theory (DFT) calculations of these activation energies.
is very large for small R but decreases, if R grows. This effect is also expected for different alloys and island structures. The pinning described in this paper depends crucially on the increase of activation energies due to an impurity nearby, such as already calculated by Bevan et al. [18] in a different context. For which alloys the effect will occur can be predicted by density functional theory (DFT) calculations of these activation energies.
For future work it would be interesting to investigate Ag islands with more than one Cu impurity. This is possible with our extended SLKMC model, since it is general enough to consider more than just one Cu atom and is only limited by the computation time to calculate new activation energies. In addition, it is possible to change the used potential and investigate the drift of different alloys. Another direction, in which progress seems possible, is to generalize existing phenomenological theories [14,15] in order to include the position of the impurity relative to the island edge and its influence on the temporal evolution.
Acknowledgments
Helpful discussions with L. Brendel are gratefully acknowledged.