
Abstract
Energetic ions lose some of their kinetic energy when interacting with electrons or nuclei in matter. Here, we discuss combined experimental and theoretical studies on such impulse driven reactions in polycyclic aromatic hydrocarbons (PAHs), fullerenes, and pure or mixed clusters of these molecules. These studies show that the nature of excitation is important for how complex molecular systems respond to ion/atom impact. Rutherford-like nuclear scattering processes may lead to prompt atom knockout and formation of highly reactive fragments, while heating of the molecular electron clouds in general lead to formation of more stable and less reactive fragments. In this topical review, we focus on recent studies of knockout driven reactions, and present new calculations of the angular dependent threshold (displacement) energies for such processes in PAHs. The so-formed fragments may efficiently form covalent bonds with neighboring molecules in clusters. These unique molecular growth processes may be important in astrophysical environments such as low velocity shock waves.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Energetic processing of complex molecular systems by photons, electrons, or heavy particles such as atoms or ions, gives rise to a rich variety of phenomena of importance for our understanding of, e.g., biological radiation damage at the nanoscale [1], the formation and degradation of aerosols in the atmosphere [2–4], the origin and evolution of molecules in space [5], and defect formation and annealing for tailoring material properties [6]. Following such interactions, the molecules may lose their excess energy by an intricate balance between emission of electrons, photons, atoms or molecules. These cooling processes are most often statistical, or thermally driven, in the sense that they occur after the energy has been distributed among all degrees of freedom. Fragmentation will then predominantly proceed through the lowest energy dissociation pathways on picosecond timescales or longer depending on the molecular heat capacities and dissociation energies. In these cases, the amount of excitation energy and not the nature of the excitation determines the fate of the molecules.
For ion/atom impact, the molecules may be weakly heated in distant electron transfer collisions, or more strongly heated when the projectile loses kinetic energy in close interactions with the electron cloud and nuclei (see figure 1). The latter effect depends strongly on the projectile charge state and energy (velocity and mass), as illustrated by the example shown in figure 2. This figure shows calculations of the energy loss per unit length (stopping power) for a He atom colliding with a pyrene (C16H10) solid [7, 8], which may be used to model interactions with (infinitely) large pyrene clusters. High-energy collisions will, initially, mostly lead to electronic excitations (the red curve 'electronic stopping' dominates in this energy region), while Rutherford-like nuclear scattering processes—referred to as 'nuclear stopping'—dominate at low collision energies (black curve). Thus, by tuning the collision energy it is possible to change the relative importance of electronic and nuclear stopping processes, and to mimic conditions in, e.g., astrophysical environments (see figure 2).

Figure 1. Schematics showing collisions between keV atomic ions and the PAH molecule coronene (C24H12). Electrons are captured at large distances leaving the so formed molecular ions only weakly heated in large impact parameters collisions ( ), while frontal collisions (
), while frontal collisions ( ) may lead to substantial heating in closer interactions with the molecular electron cloud and nuclei.
) may lead to substantial heating in closer interactions with the molecular electron cloud and nuclei.
Download figure:
Standard image High-resolution image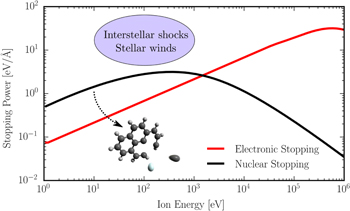
Figure 2. Nuclear and electronic stopping power for interactions between neutral He and solid C16H10 calculated using SRIM (Stopping and Range of Ions in Matter) software [7, 8]. Nuclear stopping dominates in low energy collisions while electronic stopping dominates at higher energies.
Download figure:
Standard image High-resolution imageNuclear stopping may lead to billiard ball-like interactions in which individual atoms are knocked out directly in the collision (see inset in figure 2). Here, we refer to these processes as non-statistical fragmentation processes since the energy is not distributed across all degrees of freedom before the molecule fragments. Atom knockout requires a certain threshold energy, which depends on the target atom type and its local chemical environment (i.e. bonding situation). For solids, this intrinsic property is known as the threshold displacement energy (Tdisp) and is defined as the kinetic energy a single atom, the so-called primary knock-on atom (PKA), must have to be permanently displaced from its lattice position to an interstitial site, forming a so-called Frenkel pair [9].
Such defects may introduce new properties in the materials [6]. Graphene vacancies are known to be magnetic [10] and highly reactive sites [11], while vacancies in single walled nanotubes can be used to manipulate their electrical conductivities [10]. For atom knockout in isolated molecules, the displacement energy may be defined as the energy lost by the projectile at the threshold [12, 13]. This relies on the projectile only interacting with one target atom during the collision, which is well justified for small projectiles like electrons, H, and He, but not necessarily for heavier atomic projectiles such as Xe [14].
Here we will discuss the interplay between statistical and nonstatistical fragmentation processes when polycyclic aromatic hydrocarbons (PAHs), fullerenes, and pure or mixed clusters of these molecules are impacted by ions or atoms. A few examples of these complex molecular systems are shown in figure 3. PAHs consist of fused aromatic rings, often in planar hexagonal structures, with lowest dissociation energies of about 5–8 eV for H-loss, H2-loss, and C2H2-loss [15, 16]. Calculations of PAH-cluster geometries suggest that the lowest energy structures for small neutral clusters are 1D stacks, while larger clusters form more complex 3D structures [17–19]. The cluster dissociation energies increase with increasing PAH size (number of aromatic rings) from about 0.3 eV for anthracene (C14H10) clusters [17] to about 1 eV for coronene clusters (C24H12) [18, 19].
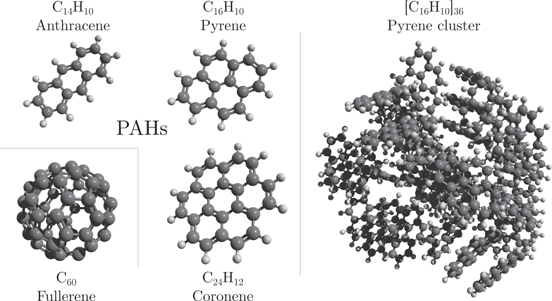
Figure 3. Examples of molecular structures for closed cage fullerenes (C60), polycyclic aromatic hydrocarbons (PAHs) and a weakly bound PAH cluster here exemplified by a cluster of 36 pyrene molecules.
Download figure:
Standard image High-resolution imagePAHs are widespread air pollutants [20, 21] and are believed to be responsible for the broad IR emission features of many astronomical objects [22–24]. Energetic processing of PAHs has been suggested as a possible route to the formation of fullerenes in space [25, 26]. Fullerenes (C60 and C70, see figure 3) have been detected in a young planetary nebula based on their characteristic IR fingerprints [27] and  ions have recently been identified as one of the carriers of the diffuse interstellar bands [28, 29]. In the laboratory, inherent fullerene properties have been extensively studied since their initial discovery in 1985 by Kroto et al [30]. It is now well established that they fragment by emission of C2-units when internally heated by photons, electrons, or atom/ions (see e.g. [31, 32] and references therein). The dissociation energy for this channel is 10.8 ± 0.3 eV for C60 [33], 9.8 ± 0.1 eV for
ions have recently been identified as one of the carriers of the diffuse interstellar bands [28, 29]. In the laboratory, inherent fullerene properties have been extensively studied since their initial discovery in 1985 by Kroto et al [30]. It is now well established that they fragment by emission of C2-units when internally heated by photons, electrons, or atom/ions (see e.g. [31, 32] and references therein). The dissociation energy for this channel is 10.8 ± 0.3 eV for C60 [33], 9.8 ± 0.1 eV for  [34], and slightly lower for neighboring fullerene sizes [34]. These experimental values agree well with those from molecular structure calculations [35]. Clusters of fullerenes typically have structures based on Mackey icosahedral geometries [36–38] with dissociation energies of about 0.3 eV [39].
[34], and slightly lower for neighboring fullerene sizes [34]. These experimental values agree well with those from molecular structure calculations [35]. Clusters of fullerenes typically have structures based on Mackey icosahedral geometries [36–38] with dissociation energies of about 0.3 eV [39].
Statistical emission of single carbon atoms from PAHs or fullerenes are associated with prohibitively high reaction barriers and are therefore not typically observed [40]. Thus, single carbon loss may be used as a fingerprint for prompt non-statistical fragmentation in the form of single atom knockout. PAHs are perhaps the most ideal systems for studying such processes due to their two dimensional rigid structures. This gives a high probability for single atom knockout and a low probability for the PKA to be recaptured at a different site in the molecule or to induce secondary knockout fragmentation. Such effects are likely to be much more important for the three-dimensional fullerenes. This makes it, as we will see, much harder to identify knockout processes in these systems.
In this topical review, we give the reader an overall picture of impulse driven reactions in fullerenes, PAHs, and their clusters, with the main focus on knockout by atoms or atomic ions. In section 2 we discuss experimental techniques for studies of low and high energy collisions, which we define as the regions in which nuclear and electronic stopping processes dominate, respectively. Section 3 is devoted to descriptions of collision models for distant and close collisions, molecular dynamics (MD) simulations, and molecular structure calculations. In section 4 we briefly review experimental and theoretical results on thermally driven reactions in isolated molecules and clusters. Studies of knockout processes in isolated molecules are discussed in section 5, where we also present new MD results on angular dependent displacement energies for PAHs. We show in section 6 that knockout processes yield highly reactive fragments similar to those reported for defect graphene. There we also discuss how these may efficiently form covalent bonds with other fragments and/or intact molecules inside clusters. Finally, in section 7 we give some concluding remarks and discuss future challenges.
2. Experimental techniques
Mass spectrometry has been the workhorse for studies of collisions involving PAHs, fullerenes and their clusters. It has been applied in essentially two different ways. One is to produce keV ion beams of these molecular systems and let them collide with neutral (atomic) targets. The center-of-mass energy is then often less than 1 keV, depending on the target mass. Alternatively, keV atomic ion beams collide with neutral PAH or fullerene targets. The center-of-mass energy is then close to the kinetic energy of the atomic projectile and thus typically much higher than in the former case. These complementary techniques may thus be used to cover the whole collision energy range shown in figure 2.
2.1. Low energy collisions
In figure 4 we show the setup used for studies of PAH+/ + He/Ne/Ar/Xe collisions at the DESIREE facility in Stockholm [40, 42, 43]. Briefly, the molecular ions are produced in an electrospray ion source using the sample preparation method of Maziarz et al [44]. The ions are then collected by a radio-frequency ion funnel, mass selected by a quadrupole mass filter, accelerated, and guided through a 4 cm long collision cell containing the target gas (He/Ne/Ar/Xe). After the collision cell, the intact and fragment ions are analyzed by means of a cylindrical lens and two pairs of electrostatic deflector plates, and recorded by a position-sensitive microchannel plate (MCP) detector. This setup allows for measurements of fragment mass spectra and absolute total destruction cross sections through attenuations of the intact ion beams as functions of the target gas pressure in the cell [40, 42].
+ He/Ne/Ar/Xe collisions at the DESIREE facility in Stockholm [40, 42, 43]. Briefly, the molecular ions are produced in an electrospray ion source using the sample preparation method of Maziarz et al [44]. The ions are then collected by a radio-frequency ion funnel, mass selected by a quadrupole mass filter, accelerated, and guided through a 4 cm long collision cell containing the target gas (He/Ne/Ar/Xe). After the collision cell, the intact and fragment ions are analyzed by means of a cylindrical lens and two pairs of electrostatic deflector plates, and recorded by a position-sensitive microchannel plate (MCP) detector. This setup allows for measurements of fragment mass spectra and absolute total destruction cross sections through attenuations of the intact ion beams as functions of the target gas pressure in the cell [40, 42].
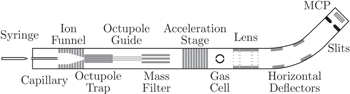
Figure 4. Schematic of the experimental setup used for studies of PAH+/ + He/Ne/Ar/Xe collisions (center-of-mass energies between about 10 eV and 1 keV) at the DESIREE facility in Stockholm, Sweden. Reprinted figure with permission from [41]. Copyright 2015 by the American Physical Society.
+ He/Ne/Ar/Xe collisions (center-of-mass energies between about 10 eV and 1 keV) at the DESIREE facility in Stockholm, Sweden. Reprinted figure with permission from [41]. Copyright 2015 by the American Physical Society.
Download figure:
Standard image High-resolution imageThe center-of-mass collision energies in these experiments are typically in the sub keV range, which is ideal for studies of nuclear stopping (knockout) driven reactions (see figure 2). In Aarhus, a similar setup was used in pioneering experiments demonstrating knockout processes in fullerenes [14] and more recently in collision induced dissociation studies of other complex molecular systems such as biomolecules [45].
2.2. High energy collisions
The first studies of keV ion impact on isolated PAH molecules were carried out using the setup shown in figure 5 [46]. There, H+ and He2+ ions were produced in separate experiments from the electron cyclotron resonance (ECR) ion source located at the ZernikeLEIF facility (KVI Groningen). The ion beam was in the 4–40 keV range, and chopped with a typical repetition rate of 10 kHz and a pulse length of about 10 ns. The so formed ion beam pulse was guided to the collision chamber to interact with a neutral anthracene target, which was evaporated from a resistively heated oven at about 360 K. This temperature was kept as high as possible to yield sufficient target densities without dissociating the PAH molecules. The intact and fragment anthracene ions produced in the collisions were extracted through a diaphragm and a lens system and analyzed by means of a reflectron time-of-flight (TOF) spectrometer equipped with an MCP detector [46].

Figure 5. Schematic of the setup used for keV atomic-ion impact on isolated PAH molecules at the ZernikeLEIF facility (KVI Groningen), the Netherlands. Figure from [46], reproduced by permission of the AAS.
Download figure:
Standard image High-resolution imageSimilar techniques have been used by other groups for studies of PAHs [47–53]. One particularly powerful technique is called Collision-Induced Dissociation under Energy Control (CIDEC) and was developed at the University of Lyon 1 [48]. This technique is based on anion formation by double electron capture in inelastic collisions between a singly charged atomic projectile and a neutral molecular (PAH) target, and relies on that there is only a single bound state in the anion. By measuring the kinetic energy loss of the anion and subtracting the energy defect of the collision, the internal energy of the molecular ion may be determined. This, together with multi-coincidence detection of ions and electrons allows for characterizing fragmentation processes in unprecedented detail [48].
In figure 6 we show the mass spectrometer setup at the ARIBE facility in Caen, which has been used for studies of PAHs, fullerenes and their clusters [47, 52, 55–58]. The latter are formed in a cluster aggregation source in which isolated molecules are evaporated from an oven and cooled by a He buffer gas at liquid nitrogen temperatures (see figure 6). This allows formation of weakly bound neutral clusters in a broad log-normal size distribution [54]. The neutral cluster targets interact with a pulsed ion beam in a similar fashion as in the Groningen experiments (see figure 5), but the collision product ions are here analyzed by means of a linear Wiley–McLaren type spectrometer terminated by a metal conversion plate. Secondary electrons from the conversion plate are guided by a weak magnetic field to a microchannel plate detector. This detection scheme gives close to unit efficiency which is crucial for coincidence measurements of all charged particles formed in a single collision event [59].

Figure 6. Schematic of the cluster aggregation source and the mass spectrometer setup at the ARIBE facility in Caen, France [54]. The ability to use two individually heated ovens allows for the productions of mixed molecular cluster beams. Reprinted figure with permission from [55]. Copyright 2014 by the American Physical Society.
Download figure:
Standard image High-resolution image3. Theoretical tools
Here, we discuss theoretical approaches which have been successfully used to describe different aspects of the collisions (see figure 1). For the sake of completeness, we will first briefly describe models for charge transfer where the molecules are only weakly heated in distant collisions, and then focus more in detail on models for penetrating collisions where the molecules are strongly heated and where atoms may be knocked out.
3.1. Distant collisions
In 1994, Walch et al reported the first experimental study of collisions between highly charged ions and C60 fullerenes [60]. This work has been followed by a vast number of studies of keV ion impact on isolated or weakly bound clusters of fullerenes (see e.g. [31, 32] and references therein), and more recently on PAHs. It is well-established through these studies that electrons are captured at large distances such that multiply charged intact fullerene/PAH ions may be formed sufficiently cold to survive on the experimental microsecond timescales. The cross sections for the electron transfer processes may be rationalized by means of the classical over-the-barrier model. In this model an electron is allowed to be transferred from the target molecule to the (multiply) charged ion when the Stark-shifted binding energy of the electron equals the maximum of the potential energy barrier experienced by the electron as it moves between the target and the projectile. This simple approach has been successfully used for atomic ions interacting with atoms [61–63], metal and insulator surfaces [64, 65], isolated fullerenes or PAHs and clusters of fullerenes or PAHs [66–68], as well as for fullerene-fullerene [69] and fullerene-surface collisions [70, 71]. In the latter models, the individual fullerene and PAH molecules are described as metal spheres [69] and infinitely thin metal disks [68], respectively. This gives a good representation of the heights and the positions of the barriers as concluded from comparisons with Density Functional Theory (DFT) calculations [68, 72]. For PAHs, it was demonstrated that it is important to take orientation dependent polarizabilities into account, in particular for large systems [68].
To gain further insights into distant collisions dynamics, quantum chemical approaches are required. A full quantum chemical treatment of the collision is computationally too demanding for current computer technology, but quasi-molecular approaches have been used for He2+ + C60 [73] and C6+ + C60 collisions [74]. There, the electronic structure of C60 was described using an extension of the spherical jellium model by Puska and Nieminen [75]. Non-spherically symmetric systems such as PAHs are considerably more challenging, and have to our knowledge not been treated in this fashion. This would lead to a more detailed understanding of the collision dynamics and is important for testing the validity of simple models for easy calculations of, e.g., charge transfer cross sections.
3.2. Electronic and nuclear stopping models
At small impact parameters the projectile atom or ion can interact directly with the electron cloud and nuclei of the target molecule. The knockout process itself is driven by nuclear scattering processes, however at high collision energies electronic stopping is often responsible for the main part of the energy transferred (see figure 2). It is thus important to bear both types of energy transfer in mind when modeling collisions. At high energies, the collisions typically take place on timescales of fs or less, and models for electronic stopping therefore usually assume a static molecular structure during the entire collision process.
Schlathölter et al presented a model for calculating the energy transferred through electronic stopping in collisions between He+ and C60 with velocities ranging from 0.1 to 1 a.u. (center-of-mass energies in the range of 1 to 100 keV) [76]. Treating the large number of C60 valence electrons as an electron gas, they noted that the inelastic energy loss through electronic scattering scaled linearly with the velocity v [77]. This allowed them to describe the stopping power as

Here γ is a friction coefficient that depends on the density parameter  , which at any point is a function of the valence electron density n0 [76]. Values for the friction coefficient as a function of electron density in an electron gas,
, which at any point is a function of the valence electron density n0 [76]. Values for the friction coefficient as a function of electron density in an electron gas,  , have been calculated by Puska and Nieminen for a range of projectile ions and energies [78]. Using these values together with a jellium shell model for the valence electron density of C60 [75], Schlathölter et al calculated the total energy transferred in a given collision geometry by integrating the stopping power S along a straight line trajectory through the static electron cloud of C60 [76].
, have been calculated by Puska and Nieminen for a range of projectile ions and energies [78]. Using these values together with a jellium shell model for the valence electron density of C60 [75], Schlathölter et al calculated the total energy transferred in a given collision geometry by integrating the stopping power S along a straight line trajectory through the static electron cloud of C60 [76].
This approach was expanded upon by Postma et al in their study of collisions between protons or alpha particles and anthracene (C14H10) at keV energies [46]. Following the scheme outlined by Schlathölter et al [76] for calculating the electronic stopping, Postma et al used Density Functional Theory to calculate the valence electron density of anthracene [46]. This approach of using quantum chemical methods to obtain the electronic density distribution may be applied on a wide range of molecular systems. Postma et al subsequently utilized a Monte Carlo approach to calculate the electronic stopping energy distribution for randomly selected collision trajectories between the ions and the anthracene target at different orientations [46]. The top panel of figure 7 shows the electronic stopping energy distributions for  randomly distributed straight-line trajectories of He (
randomly distributed straight-line trajectories of He ( a.u.,
a.u.,  keV) colliding with anthracene face-on and edge-on (see insets in figure 7) [46]. The energy distribution from randomly oriented molecules is shown in the lower panel of figure 7.
keV) colliding with anthracene face-on and edge-on (see insets in figure 7) [46]. The energy distribution from randomly oriented molecules is shown in the lower panel of figure 7.

Figure 7. Calculated distributions of energy deposited through electronic stopping by He traversing a single anthracene molecule along straight face-on (blue curve) and edge-on (red curve) trajectories (top panel). Results from the same types of calculations but with random orientation of the anthracene molecule are shown in the lower panel. Figure from Postma et al [46], reproduced by permission of the AAS.
Download figure:
Standard image High-resolution imageA similar method was used to study the interactions between 50–180 keV He+ or He2+ ions and naphthalene (C10H8) by Mishra et al [53]. Using DFT to calculate the valence electron density of the target molecules, they then determined the stopping power using the Local Density Approximation (LDA) model developed by Lindhard et al for swift low charge state ions interacting with an electron gas [79, 80].
At low center-of-mass collision energies (ECM less than about 1 keV for H and He projectiles, see figure 2) nuclear stopping is the dominant mechanism for energy transfer. Nuclear scattering is the result of the projectile and target nuclei repelling each other through Coulomb interactions. This short range interaction can be described as binary collisions between a projectile ion and individual atoms in the target as

where Zp and Zi are the atomic numbers of the projectile and the i individual target atoms, and  is a screening function for describing the combined shielding effect of the electrons surrounding the projectile and target nuclei.
is a screening function for describing the combined shielding effect of the electrons surrounding the projectile and target nuclei.
The screened Bohr potential [81] and the Ziegler–Biersack-Littmark (ZBL) potential [7] are two potentials where such a screening function has been parameterized for a wide range of colliding atom/ion pairs. In both cases the screening function is a relatively simple function of the internuclear distance rpi. In the screened Bohr potential the screening function is described as a power-law that depends on  , and Zi [81]. The ZBL potential uses a screening function of the form
, and Zi [81]. The ZBL potential uses a screening function of the form
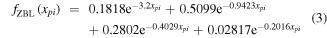
where
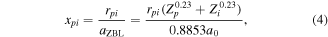
aZBL is the so-called screening length, and a0 is the Bohr radius [7].
Chen et al [82] used a similar model as Postma et al [46] for electronic stopping together with potentials for the nuclear stopping (ZBL and screened Bohr) to study ion-PAH collisions. In this way they calculated the relative contributions from both of these stopping processes over a wide energy range. As a result they determined a scaling law for estimating the cross section of single C knockout from PAH molecules of different sizes [82]. Figure 8 shows the contributions of energy transfer through electronic and nuclear stopping in collisions between He and coronene (C24H12) along straight trajectories perpendicularly through the molecular plane, at center-of-mass collision energies of 11 keV (top row) and 110 eV (bottom row) [82]. At 11 keV we see that energy transfer through electronic stopping (left column) is completely dominant, with the nuclear stopping (middle panel) only significantly contributing to the total energy for trajectories where the impact parameter  for any given atom in the target molecule [82]. As can be expected, the largest contribution from electronic stopping is from regions with the highest electron density, i.e. in the bonds. On the other hand, nuclear stopping is responsible for most of the energy transfer at 110 eV collision energies, with the contribution from electronic stopping there only reaching a few eV at most [82].
for any given atom in the target molecule [82]. As can be expected, the largest contribution from electronic stopping is from regions with the highest electron density, i.e. in the bonds. On the other hand, nuclear stopping is responsible for most of the energy transfer at 110 eV collision energies, with the contribution from electronic stopping there only reaching a few eV at most [82].

Figure 8. Electronic and nuclear stopping energy distributions as calculated by Chen et al [82] for face-on collisions between He and coronene at 11 keV (top row) and 110 eV (bottom row) center-of-mass energies. The right column shows the total stopping energy distributions, i.e. the sums of the electronic and nuclear stopping contributions. Reprinted with permission from [82]. Copyright 2014, AIP Publishing LLC.
Download figure:
Standard image High-resolution image3.3. Molecular dynamics simulations of collisions and bond-forming reactions
In the stopping calculations discussed in the previous section, the nuclear and electronic stopping energies are both calculated for straight projectile trajectories through a target molecule with a static structure. For calculations of the electronic stopping, where the electronic density of the target is required, performing fully dynamic quantum chemical calculations of collisions involving larger molecular systems comes with a high computational cost. Nonetheless, there are models that combine classical molecular dynamics (MD) with nonadiabatic quantum chemical calculations to model collisions over a wide range of energies [83]. Nuclear stopping models on the other hand are well suited for implementation in purely classical MD simulations when using functions like the ZBL [7] or screened Bohr [81] potentials to describe these interactions. Classical MD simulations are for this reason particularly useful when modeling low center-of-mass energy collisions, in the energy range where energy transfer through nuclear stopping dominates over electronic stopping.
A number of classical force fields (also known as potentials) have been used to describe intramolecular bonds in simulations of impulse driven fragmentation and bond formation. A class of force fields that are frequently used for these types of simulations are the reactive many-body force fields. Examples of these are the reactive potential by Tersoff [84, 85], the two generations of the REactive Bond Order (REBO) potential by Brenner et al [86, 87], and the Adaptive Intermolecular REBO (AIREBO) potential by Stuart et al [88], which is an extension of the REBO force field that includes a description of dispersion forces. What separates these force fields from more simplistic two-body potentials, such as harmonic or Morse potentials, is in how they dynamically adjusts the bond strengths as functions of the (changing) positions of all atoms in the system during a simulation. All of these potentials are developed and parameterized for systems containing carbon and allow the C atoms to form either sp-, sp2-, or sp3-type bonds in a realistic manner depending on the number and type of the neighboring atoms. This means that bond cleavage and the formation of new bonds can be modeled dynamically using classical MD tools.
While the reactive force fields used in classical MD simulations allow for dynamic bond breakage and formation, they do not include any charge effects. The charge distribution of a molecule or molecular ion depends on how the electron density distribution changes as atoms move and bonds are rearranged. In classical MD, partial charges are generally assigned statically to atoms or massless dummy atoms when they are considered important. In the case of fullerene or PAH molecules, which are large systems with many delocalized electrons, the removal of a single electron has little effect on the stability and reactivity of the molecule [82, 89]. The classical simulations discussed in this review, which do not included charges, are thus used to study both neutral and singly charged systems.
Besides being used to model collisions and prompt knockout processes, MD simulations are also applied in studies of secondary processes such as statistical fragmentation [41, 76] and bond-forming reactions inside molecular clusters [55, 57, 58, 89].
3.4. Quantum chemical calculations
Beyond the use of classical force fields to describe interactions and molecular structures in MD simulations, quantum chemical calculations play a crucial role in our understanding of impulse driven reactions. The computational cost of high level quantum chemical methods, e.g. DFT, and the large PAH and fullerene systems limit the use of such calculations mainly to molecular structure and quantum-state energy calculations. Nonetheless, the ability to accurately determine characteristics of molecules and fragments is important.
Parameterized and semi-classical quantum chemical methods can in some cases be a useful alternative to full DFT methods, offering many of the advantages of higher level theories, but at a much reduced computational cost. While this reduction does generally lead to lower accuracies, they still provide very useful approximate results for many systems. One such method that has been used to study PAHs and fullerenes is the self consistent charge density functional tight binding (SCC-DFTB) method [90, 91]. SCC-DFTB is a parameterized simplification of DFT and has a much lower computational cost [90, 91]. This means that SCC-DFTB can be used to perform MD simulations that are only slightly more expensive than the classical ones, while they also account for additional effects such as charge dynamics.
Specific examples of the use of quantum chemical structure calculations are those determining the stability [40] and reactivity [57, 82] of knockout products, identifying reaction barriers and dissociation energies for molecules and molecular clusters [41, 52, 92, 93]. The results may be used for benchmarking or as input parameters for simpler theoretical models [89], such as positions of nuclei and electronic densities for use in stopping models (see section 3.2) [46, 53, 82].
4. Statistical fragmentation of molecules and clusters
4.1. Isolated fullerenes and PAHs
Statistical fragmentation of fullerenes has been extensively studied in the laboratory following interactions with ions/atoms. A comprehensive review of that field is beyond the scope of the present work and for such aspects we refer the reader to [31, 32]. It is well established through these studies that internally hot fullerenes fragment by loss of C2-units, sometimes in long evaporation sequences, depending on the energy deposited in the collisions. This is illustrated in figure 9, which shows a mass spectrum from 200 keV  + H2 (
+ H2 ( eV) collisions [94]. To the left of the intact ion peak (
eV) collisions [94]. To the left of the intact ion peak ( ) there is a distribution of peaks separated by two carbon masses. These are due to statistical fragmentation processes in which the molecules are significantly heated and where fragmentation predominantly proceeds through the lowest energy dissociation pathways (C2-emission),
) there is a distribution of peaks separated by two carbon masses. These are due to statistical fragmentation processes in which the molecules are significantly heated and where fragmentation predominantly proceeds through the lowest energy dissociation pathways (C2-emission),

A corresponding fragment mass distribution with the same pattern for doubly charged fullerenes is seen at lower mass-to-charge ratios in figure 9.

Figure 9. Mass spectrum due to 200 keV  + H2 collisions, showing that fullerenes cool down by emission of C2-units in statistical fragmentation processes. Reprinted figure with permission from [94]. Copyright 1992 by the American Physical Society.
+ H2 collisions, showing that fullerenes cool down by emission of C2-units in statistical fragmentation processes. Reprinted figure with permission from [94]. Copyright 1992 by the American Physical Society.
Download figure:
Standard image High-resolution imagePAHs have been much less studied than fullerenes by ion/atom impact, in particular in the keV collision energy regime [46–51, 53]. Postma et al reported the first keV ion-PAH collision study in which H+/He2+ ions collided with anthracene (C14H10). In figure 10 we show their mass spectrum for 30 keV He2+ + C14H10 collisions [46]. There are strong peaks corresponding to singly ( ) and doubly charged (
) and doubly charged ( ) intact ions. These stem from distant electron transfer collisions in which small amounts of energy are deposited (see section 3.1). To the left of the C14
) intact ions. These stem from distant electron transfer collisions in which small amounts of energy are deposited (see section 3.1). To the left of the C14 and C14
and C14 peaks there are less intense peaks corresponding to H- and C2H2-losses (inset in figure 10):
peaks there are less intense peaks corresponding to H- and C2H2-losses (inset in figure 10):
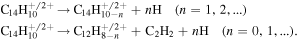
These are the lowest energy dissociation pathways for neutral and moderately charged PAHs (see e.g. [15, 16]). The rich distributions of small hydrocarbon ions may be attributed to multi-fragmentation processes following frontal collisions in which large amounts of energy are transferred mainly in electronic stopping processes (see figure 2). Using a simple collision model described in section 3.2, Postma et al [46] found that the electronic stopping is almost equal for 30 keV He2+ and 10 keV H+ impact, still markedly different fragmentation patterns were observed in these two cases. The differences were attributed to double electron capture by the He2+ projectiles, a channel which is strongly suppressed for protons [46].

Figure 10. Mass spectrum due to 30 keV He2+ + C14H10 collisions. The zoom-in shows the mass region between the peaks for singly ( ) and doubly charged (
) and doubly charged ( ) intact C14H10. Figure from [46], reproduced by permission of the AAS.
) intact C14H10. Figure from [46], reproduced by permission of the AAS.
Download figure:
Standard image High-resolution imageŁawicki et al [47] investigated the effect of the projectile charge state in a wider range using collisions between He2+/O3+/Xe20+ and pyrene or coronene (C16H10/C24H12). Singly up to quadruply charged intact molecular ions were clearly observed with Xe20+ projectiles, demonstrating that it is possible to produce multiply charged PAHs close to their ultimate Coulomb stability limits [15] in distant electron transfer collisions. The experimental ionization yields compare favorably with those from the classical over-the-barrier model for infinitely thin circular metal disks [68].
Multiply charged PAHs open up new statistical fragmentation pathways where more than one charged fragment is emitted in Coulomb-driven processes. Reitsma et al [51] measured coincidences between charged fragments emitted from doubly charged naphthalene (C10 ) and found that the strongest fragmentation channel corresponds to C10
) and found that the strongest fragmentation channel corresponds to C10 C7
C7 + C3
+ C3 , i.e. fragments with an odd number of carbons. The results were successfully interpreted in view of molecular structure calculations of dissociation energies and reaction barriers [51]. The kinetic energy releases in these processes are in the 2–3 eV range, which could be important for overcoming low barriers in molecular growth processes that may occur in, e.g., the interstellar medium [51].
, i.e. fragments with an odd number of carbons. The results were successfully interpreted in view of molecular structure calculations of dissociation energies and reaction barriers [51]. The kinetic energy releases in these processes are in the 2–3 eV range, which could be important for overcoming low barriers in molecular growth processes that may occur in, e.g., the interstellar medium [51].
The group in Lyon used the CIDEC technique (see section 2.2) to measure the internal energies associated with different fragmentation pathways for doubly charged anthracene [48, 49]. The results from one of these studies are shown in figure 11. The 2D-images in panel (a) display coincidence measurements of the ion mass-to-charge ratio (vertical axis) and the voltage applied to analyze the anions formed in F+ + C14 F− + C14
F− + C14 collisions (horizontal axis). The analyzer voltage is used to determine the kinetic energy loss of the F− projectile (
collisions (horizontal axis). The analyzer voltage is used to determine the kinetic energy loss of the F− projectile ( ), and then the excitation energy (
), and then the excitation energy ( ) of the intact or fragmented C14
) of the intact or fragmented C14 ion through the relation
ion through the relation

where  is the internal energy of the neutral anthracene and
is the internal energy of the neutral anthracene and
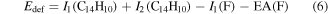
is the energy defect. In [48] Martin et al used  from literature values of the first and second ionization energies for anthracene (I1(C14H10) and I2(C14H10)), and the first ionization energy (I1(F)) and electron affinity (EA(F)) for fluorine. The panels to the left in figure 11 show the projections for four different 2D-spots on the excitation energy axis (see the bottom scale). From top to bottom (b–e) the distributions have maxima at 8.5 eV (intact ion), 13.8 eV (loss of two hydrogens), 10.4 eV (C2H2-loss), and 14.3 eV (loss of two C2H2). Thus about 10 eV is required to induce fragmentation on the experimental microsecond timescales. This is significantly higher than the lowest dissociation energy pathway (4.3 eV for C2H2-loss [49]), which reflects the large heat capacity of PAHs. They will thus eventually fragment unless there are sufficiently fast competing (radiative) cooling processes, as observed for singly charged anthracene in the small electrostatic ion storage ring, MINIring, in Lyon [96].
from literature values of the first and second ionization energies for anthracene (I1(C14H10) and I2(C14H10)), and the first ionization energy (I1(F)) and electron affinity (EA(F)) for fluorine. The panels to the left in figure 11 show the projections for four different 2D-spots on the excitation energy axis (see the bottom scale). From top to bottom (b–e) the distributions have maxima at 8.5 eV (intact ion), 13.8 eV (loss of two hydrogens), 10.4 eV (C2H2-loss), and 14.3 eV (loss of two C2H2). Thus about 10 eV is required to induce fragmentation on the experimental microsecond timescales. This is significantly higher than the lowest dissociation energy pathway (4.3 eV for C2H2-loss [49]), which reflects the large heat capacity of PAHs. They will thus eventually fragment unless there are sufficiently fast competing (radiative) cooling processes, as observed for singly charged anthracene in the small electrostatic ion storage ring, MINIring, in Lyon [96].

Figure 11. CIDEC measurements of F+ + C14 F− + C14
F− + C14 collisions (see text). (a) 2D images from coincidence measurements of the mass-to-charge ratio of the intact or fragmented C14
collisions (see text). (a) 2D images from coincidence measurements of the mass-to-charge ratio of the intact or fragmented C14 (vertical axis) and the voltage applied to determine the kinetic energy loss of F− (horizontal axis). (b)–(e) Projections on the excitation energy (internal energy) axis for the four 2D-spots assigned to, from top to bottom: the intact ion (
(vertical axis) and the voltage applied to determine the kinetic energy loss of F− (horizontal axis). (b)–(e) Projections on the excitation energy (internal energy) axis for the four 2D-spots assigned to, from top to bottom: the intact ion ( ), loss of two hydrogen atoms (
), loss of two hydrogen atoms ( ), C2H2-loss (
), C2H2-loss ( ), and loss of two C2H2 molecules (
), and loss of two C2H2 molecules ( ). The top scale and the bottom scale show the kinetic energy loss, ΔE, for F− and the excitation energy (internal energy) of the parent C14
). The top scale and the bottom scale show the kinetic energy loss, ΔE, for F− and the excitation energy (internal energy) of the parent C14 ion, respectively. Reprinted figure with permission from [48]. Copyright 2012 by the American Physical Society.
ion, respectively. Reprinted figure with permission from [48]. Copyright 2012 by the American Physical Society.
Download figure:
Standard image High-resolution imageOne interesting aspect in this context is whether molecular hydrogen may be formed from internally heated native PAHs [16, 48, 93, 97]. This has been the subject of debate as this channel is impossible to distinguish from sequential emission of two hydrogen atoms (H + H) by means of mass spectrometry alone. A recent combined experimental and theoretical study suggests that keV ions may effectively induce H2-formation from PAHs, but that the same process is not possible through absorption of single photons with energies below the Lyman limit [93]. However, this situation needs to be unambiguously clarified through additional experiments.
4.2. Clusters: evaporative cooling and coulomb heating
To the best of our knowledge, all experimental studies of keV ion impact on weakly bound PAH and fullerene clusters have so far been carried out at the ARIBE facility in Caen (see section 2.2). A recent review of this work is given in [98] and, there, studies of biomolecules, fullerenes, and PAHs are also included. We will now highlight some of the studies on PAHs and fullerenes that were performed at this facility [92, 95, 99, 100, 100].
In 2003, Manil et al [99] reported on the first measurements of keV ions (in this case Xe25+) interacting with clusters of fullerenes. In contrast to previous results on the ionization of weakly bound atomic rare gas clusters [101], they found that the charge is rapidly redistributed among all individual fullerenes in the cluster. It was later shown that this charge communication is ultrafast and takes place on subfemtosecond timescales [59, 100, 102]. Further, Manil et al [99] showed that the excitation energy is rapidly shared among the cluster constituents such that the individual molecules become protected from damage in a surrounding environment.
Rapid charge and energy distribution have also been observed for clusters of e.g. biomolecules [103–106] and PAHs [92, 95]. In figure 12, we show an example for 11.25 keV He+ and 360 keV Xe20+ ions colliding with anthracene monomers and clusters [95]. In the rightmost panels of figure 12, we show the mass region for intact monomers and clusters, which display decreasing intensity distributions as functions of cluster size for both projectiles. These distributions are rather similar but stem from markedly different processes. For He+, the clusters are mainly heated through electronic stopping of ions interacting closely with a few of the molecular building blocks. The excess energy is rapidly shared among the cluster constituents and the cluster cool by the evaporation of neutral anthracene monomers. This results in singly charged intact PAH monomers which are significantly colder than for collisions with a single anthracene molecule isolated in vacuum. As a consequence, the monomer fragmentation yield reduces by a factor ten for collisions with clusters compared to monomers, as shown in the middle and left upper panels in figure 12, respectively. Thus, the individual molecules are protected from damage by the surrounding (cluster) environment [95].

Figure 12. Mass spectra for 11.25 keV He+ (upper panels) and 360 keV Xe20+ ions (lower panels) colliding with anthracene monomers (C14H10) and clusters ([C14H10]k). Left panels: collisions with monomer targets. Middle panels: monomer fragment region for collisions with clusters. The insets show the relative Cn intensity distributions as functions of n for collisions with clusters (full symbols) and monomers (open symbols). Right panels: cluster fragment distributions where the insets show zoom-ins in logarithmic intensity scales. Reprinted figure with permission from [95]. Copyright 2010 by the American Physical Society.
intensity distributions as functions of n for collisions with clusters (full symbols) and monomers (open symbols). Right panels: cluster fragment distributions where the insets show zoom-ins in logarithmic intensity scales. Reprinted figure with permission from [95]. Copyright 2010 by the American Physical Society.
Download figure:
Standard image High-resolution imageFor Xe20+ (lower panel in figure 12), the collisions are dominated by distant electron transfer processes and small amounts of energy transfers directly in the collisions (see section 4.1). In this case, the monomers are not protected from damage as the fragmentation yields in the lower middle panel of figure 12 are similar to the ones for a target of monomers (lower left panel). One possible explanation is heating by Coulomb explosions of multiply charged clusters following rapid charge redistribution. In such cases a substantial part of the potential energy is converted into internal energy of emitted singly charged monomers [95]. This effect has been shown to be important for multiply charged fullerene dimers [59, 100].
5. Nonstatistical fragmentation of fullerenes and PAHs
At low center-of-mass collision energies nuclear scattering processes are the dominant mechanism for energy transfer and the contributions from electronic stopping processes may be very small and close to negligible. Despite this, nuclear scattering can still deposit enough energy (about 10 eV for small PAHs [48]) for statistical fragmentation processes to be important on the microsecond timescales of experiments. There will therefore always be competition between statistical and nonstatistical fragmentation processes and the direct results of prompt knockout may be masked by subsequent statistical fragmentation. A schematic overview of this competition between statistical and nonstatistical fragmentation processes in PAHs and their different timescales is shown in figure 13 [107].

Figure 13. Overview of competing statistical and non-statistical fragmentation processes (knockout) for PAHs. The products highlighted in gray are those that, when detected in experiments, can unambiguously be determined to be formed by knockout. Figure from [107].
Download figure:
Standard image High-resolution image5.1. Knockout processes in fullerenes
The first theoretical studies of prompt single atom knockout from fullerene molecules predates the first experimental findings. In 1994, Cui et al presented a study on particle radiation damage to C60 molecules using classical MD simulations [108]. They modeled the C60 using the Tersoff potential to describe the bonds in the molecule together with the ZBL potential for close range interactions where nuclear scattering was important. Cui et al did not include projectiles in their simulations. Instead they gave the PKA a velocity at the beginning of the simulation, as if that atom had been struck by a projectile. From these simulations they predicted the knockout driven formation of C58, C59, and endohedral C@C59 in collisions between C60 and energetic particles [108]. They also deduced an average displacement energy of 29 eV for dislocating or completely removing a C atom from C60 [108].
This study was followed by work from Ehlich et al [109], Larsen et al [14], and Tomita et al [110] who used classical MD simulations to explicitly model collisions between noble gas atoms and C60. These groups all identified knockout driven fragmentation of C60 as an important mechanism in low center-of-mass energy collisions. The addition of a projectile atom to the simulations enabled them to calculate internal energy distribution for the C60 molecules and any large Cx fragments following the collision [14, 109, 110].
Experimental evidence of nonstatistical fragmentation of fullerene molecules was first reported by Larsen et al [14] and later by Tomita et al [110]. In these experiments they let C60 ions, both cations and anions, collide with nobel gas targets. However, they only directly observed knockout, i.e.  fragments, with the
fragments, with the  projectiles [14, 110]. Both the C60 anions and cations were produced using the same electrospray ionization source, but the negative ions were shown to have lower internal energies than the positive ions [14, 110]. The higher internal energy of the
projectiles [14, 110]. Both the C60 anions and cations were produced using the same electrospray ionization source, but the negative ions were shown to have lower internal energies than the positive ions [14, 110]. The higher internal energy of the  ions prior to the collisions meant that the fragments also had higher internal energies than those from
ions prior to the collisions meant that the fragments also had higher internal energies than those from  projectiles. The result of this difference was that the
projectiles. The result of this difference was that the  fragments that were formed from the collisions with
fragments that were formed from the collisions with  projectiles were more likely than those from
projectiles were more likely than those from  projectiles to survive long enough to be detected [14, 110]. Using internal energy distributions from their MD simulations, Larsen et al further calculated theoretical mass spectra that included both effects of prompt non-statistical fragmentation and the subsequent statistical fragmentation [14]. These compared well with their experimental results and showed that knockout was an important first fragmentation step in collisions with fullerenes at sufficiently low energies.
projectiles to survive long enough to be detected [14, 110]. Using internal energy distributions from their MD simulations, Larsen et al further calculated theoretical mass spectra that included both effects of prompt non-statistical fragmentation and the subsequent statistical fragmentation [14]. These compared well with their experimental results and showed that knockout was an important first fragmentation step in collisions with fullerenes at sufficiently low energies.
In addition to positively charged fragments, Tomita et al also detected  and
and  fragments produced by prompt single and double C atom knockout [110]. Figure 14 shows zoom-ins of negative ion mass spectra obtained by Tomita et al for collisions between
fragments produced by prompt single and double C atom knockout [110]. Figure 14 shows zoom-ins of negative ion mass spectra obtained by Tomita et al for collisions between  and He and Ne targets at center-of-mass energies of 276 eV and 1350 eV, respectively [110]. The detection of negatively charged fragments in the collisions shows that knockout may, under certain conditions, result in fragments with very low internal energies [110]. This is because most of the excitation energy is deposited to and carried away by the atom knocked out in the collisions. Collisions leading to more significant heating of the molecules also leads to the loss of one or more electrons, and as a result, the positively charged fragments that are detected [14, 110].
and He and Ne targets at center-of-mass energies of 276 eV and 1350 eV, respectively [110]. The detection of negatively charged fragments in the collisions shows that knockout may, under certain conditions, result in fragments with very low internal energies [110]. This is because most of the excitation energy is deposited to and carried away by the atom knocked out in the collisions. Collisions leading to more significant heating of the molecules also leads to the loss of one or more electrons, and as a result, the positively charged fragments that are detected [14, 110].

Figure 14. Mass spectra of negatively charged products from collisions between  projectiles and He (left) and Ne (right) at center-of-mass collision energies of 276 eV, and 1350 eV, respectively [110]. C59 anions are assigned to single C knockout. Reprinted figure with permission from [110]. Copyright 2002 by the American Physical Society.
projectiles and He (left) and Ne (right) at center-of-mass collision energies of 276 eV, and 1350 eV, respectively [110]. C59 anions are assigned to single C knockout. Reprinted figure with permission from [110]. Copyright 2002 by the American Physical Society.
Download figure:
Standard image High-resolution imageAround this time, Kunert and Schmidt presented a theoretical study of atomic ions colliding with C60 molecules [111]. Using a method called nonadiabatic quantum molecular dynamics (NA-QMD) [83] they combined classical MD simulations with time-dependent DFT calculations to directly and self-consistently model both electronic and vibronic excitations. In this way they studied collisions with several different projectile ions at a wide range of energies and observed knockout driven fragmentation of the fullerene molecules. This study also showed that at collision velocities above about 0.25 a.u. (equivalent to center-of-mass energies of about 6 keV, 18 keV, and 59 keV for He+, C+, and Ar+ projectile ions, respectively) the stopping models from SRIM [7, 8] overestimated the energies transferred in collisions [111].
The first report of C59 ions being directly detected from collisions with C60 cations came over a decade later from experiments at lower energies [43]. Here  ions were made to collide with He or Ne at center-of-mass energies of 50 eV and 240 eV, respectively. From collisions with He, there was no direct evidence of prompt single C knockout detected [43]. On the other hand with the Ne target there was a clear, but weak, signal of
ions were made to collide with He or Ne at center-of-mass energies of 50 eV and 240 eV, respectively. From collisions with He, there was no direct evidence of prompt single C knockout detected [43]. On the other hand with the Ne target there was a clear, but weak, signal of  in the mass spectrum [43]. Even at these low collision energies, where electronic stopping is weak [82], the fragment mass spectra from fullerenes are dominated by products from statistical fragmentation [43]. While C60 are large molecules with high dissociation energies, their three dimensional structures cause any atom that penetrates the molecules to interact with two layers of atoms. Any C atom that is knocked out by the projectile may also in itself become a secondary projectile that can inflict further damage to the rest of the molecule [43]. Furthermore, the colliding atom or the PKA (primary knock-on atom) may be captured by C60 forming an endohedral fullerene or a C@C59 molecule [108]. In the former case, the available center-of-mass energy, which may be several tens of eV, will be converted into internal energy of the the combined system [43, 111]. The latter cannot be unambiguously identified by means of mass spectrometry alone as it has the same mass-to-charge ratio as the intact C60.
in the mass spectrum [43]. Even at these low collision energies, where electronic stopping is weak [82], the fragment mass spectra from fullerenes are dominated by products from statistical fragmentation [43]. While C60 are large molecules with high dissociation energies, their three dimensional structures cause any atom that penetrates the molecules to interact with two layers of atoms. Any C atom that is knocked out by the projectile may also in itself become a secondary projectile that can inflict further damage to the rest of the molecule [43]. Furthermore, the colliding atom or the PKA (primary knock-on atom) may be captured by C60 forming an endohedral fullerene or a C@C59 molecule [108]. In the former case, the available center-of-mass energy, which may be several tens of eV, will be converted into internal energy of the the combined system [43, 111]. The latter cannot be unambiguously identified by means of mass spectrometry alone as it has the same mass-to-charge ratio as the intact C60.
5.2. Knockout processes in PAHs
Experimental mass spectra from 110 eV collisions between He and PAH cations of different sizes are shown in figure 15 [40]. Here the collision energy is low enough such that significant amounts of energy is transferred from the projectile to the target almost entirely through nuclear stopping [82] (see also figures 2 and 8). When this is the case the interactions will initially be localized to single atoms in the molecules, with relatively little overall heating. The mass spectra recorded at 110 eV center-of-mass energy are very different from those where PAH molecules are strongly heated by electronic stopping in much faster keV collisions (see e.g. the top-left panel in figure 12). There are strong mass peaks corresponding to losses of about 12 amu, which are highlighted in gray in figure 15. This is from PAH fragments where a single C atom, and possibly a number of H atoms, has been knocked out of the molecule in the collision and where the fragment has survived long enough to be detected [40].

Figure 15. Mass spectra due to collisions between PAH cations of different sizes and a He target, all at 110 eV center-of-mass energy. The peaks corresponding to direct single C (CHx-loss,  ) are highlighted in gray. Reprinted figure with permission from [40]. Copyright 2014 by the American Physical Society.
) are highlighted in gray. Reprinted figure with permission from [40]. Copyright 2014 by the American Physical Society.
Download figure:
Standard image High-resolution imageThe peaks for mass loss of about 24 amu in figure 15 are mainly due to statistical fragmentation in the form of C2Hx-loss. The corresponding events stem from close collisions where the energy transfer to the individual atoms were too small for knockout, but sufficient for statistical fragmentation on the experimental microsecond timescales. For anthracene, about 10 eV is required for statistical fragmentation (C2H2-loss etc) on the microsecond timescale [48]. Peaks due to fragments that have lost three or more C atoms, are either due to pure statistical processes, or to secondary statistical fragmentation processes following knockout of a C atom.
The intensities of the CHx-loss and C2Hx-loss peaks in figure 15 reflect the relative contributions of nonstatistical versus statistical fragmentation processes [40, 82]. For small PAH molecules like anthracene, the strongest feature in the mass spectrum is from C2Hx-loss, but when the PAH size increases the CHx-loss peak begins to dominate. This is due to the increasing heat capacities, and that the dissociation energies and the total excitation energies are rather independent of the PAH size [40, 82]. The reason for the latter is that the interactions with the electron cloud and nuclei is mainly localized along the projectile's trajectory through the PAH molecule (see figure 8). In all but the most extreme edge-on trajectories, the energy deposited in a collision with a given center-of-mass energy will thus be nearly the same regardless of PAH size. Because of this, the absolute cross sections for the knockout of a single C atom from a PAH scales with the number C atoms in the molecule, while at the same time the rate of statistical fragmentation depends on the internal temperature of the molecule or fragment after the collision (which is lower for large PAH systems for a given energy input). So not only is a large PAH less likely to fragment statistically than a small molecule, but the same is also true for fragments due to prompt C knockout [40, 82] (although there are some smaller isomeric differences [107]). Therefore in the case of very large PAH molecules—such as those believed to be abundant in the interstellar medium and with, on average, about 50–200 C atoms [24]—nonstatistical fragmentation may be an important starting point for molecular evolution [40, 82].
In figure 16 we show experimental (black) and simulated (red) absolute fragmentation cross sections for 5 keV anthracene cations colliding with He, Ne, Ar, and Xe targets from [42]. In the same figure, we also show simulated knockout fragmentation cross sections (blue). Prompt knockout is only responsible for about half of the total destruction cross section with He target atoms. The other half comes from anthracene molecules that have been heated to at least 10 eV internal energy in nuclear scattering processes that did not lead to knockout [48]. For heavier targets the contribution from non-statistical fragmentation increases—more than 80% of the total cross sections with Ne, Ar, and Xe is from such processes. With very large targets, e.g. Xe, the destruction cross section is close to the geometrical size of the molecules. This means that all penetrating collisions lead to fragmentation and the molecule is no longer partially transparent to the target atoms as is the case for e.g. He [42].
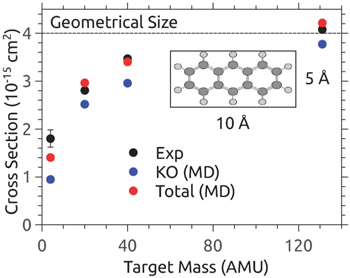
Figure 16. Absolute total fragmentation cross sections for 5 keV collisions between anthracene cations and noble gas atoms (He, Ne, Ar, and Xe) from experiments and from MD simulations. The MD-results are shown as cross sections for knockout alone (blue points), and as the sum of the knockout cross section and (delayed) statistical fragmentation from molecules with internal energies of at least 10 eV (red points). The geometrical size of the molecule is the size of the box shown in the inset multiplied by 0.8 to take the random molecular orientations of the molecules in the collisions into account. Reproduced from [42] with permission from the PCCP Owner Societies.
Download figure:
Standard image High-resolution image5.3. Threshold energies for nonstatistical fragmentation
Postma et al performed classical MD simulations to study non-statistical fragmentation in collisions of H or He atoms with anthracene, coronene, or sheets of graphene [12]. They used the ZBL potential to model nuclear scattering and the REBO potential to describe the bonds in the target systems. Using these simulations, Postma et al determined absolute cross sections for single C knockout as well as the displacement energy for the removal of a C atom from the molecules in direct face on collisions. They found that the displacement energy for removing a C atom from PAH molecules was similar to that for graphene, with a slight dependence on the location of the C atom in the PAH molecule. Further, their displacement energies had a weak dependence on the projectile species and was slightly higher for He (26.7 eV) than for H (25.5 eV) on coronene [12]. This dependence was attributed to interactions between the projectile and atoms other than the one that is knocked out from the target—with the larger He atom interacting more strongly with other atoms in the PAH than an H atom does. Overall the displacement energies determined by Postma et al [12] using classical MD simulation were only slightly larger than those determined for the removal of single C atoms from graphene by electron impact yielding results of 22.03 eV (ab initio calculations [112]) and 23.6 eV (experiments [113, 114]).
Stockett et al reported on the first measurements of the energy threshold for single carbon knockout from PAH molecules in collisions with He [13]. Cross sections for single C knockout as a function of center-of-mass collision energy from this study are shown in the top panel of figure 17. Classical MD simulations were used for comparison and showed the same energy dependence for the knockout cross section as in the experiment, but with a shift of  in the measured threshold energy. The mean MD displacement energy for anthracene, pyrene, and coronene was found to be 27.0 ± 0.3 eV, in good agreement with the values by Postma et al for anthracene [12]. However, using the experimental results to rescale the theoretical displacement energies (bottom panel of figure 17), Stockett et al deduced a semi-empirical mean displacement energy of 23.3 ± 0.3 eV for (randomly oriented) He-PAH collisions [13], which agrees well with the values for electron impact on graphene [112–114].
in the measured threshold energy. The mean MD displacement energy for anthracene, pyrene, and coronene was found to be 27.0 ± 0.3 eV, in good agreement with the values by Postma et al for anthracene [12]. However, using the experimental results to rescale the theoretical displacement energies (bottom panel of figure 17), Stockett et al deduced a semi-empirical mean displacement energy of 23.3 ± 0.3 eV for (randomly oriented) He-PAH collisions [13], which agrees well with the values for electron impact on graphene [112–114].
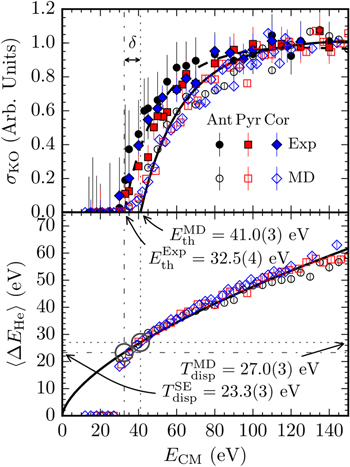
Figure 17. Top panel: normalized cross sections for single C knockout from three PAHs as functions of the center-of-mass energy in collisions with He. The lowest collision energies required for knockout (Eth) from the experiments and MD simulations are offset by  . Bottom panel: the MD-simulations give kinetic energy losses of the He atom,
. Bottom panel: the MD-simulations give kinetic energy losses of the He atom,  , at the experimental (
, at the experimental ( ) and MD (
) and MD ( ) threshold values through the circled crossing points with the fitted line. This yields Semi-Empirical (SE) and MD displacement energies of
) threshold values through the circled crossing points with the fitted line. This yields Semi-Empirical (SE) and MD displacement energies of  and
and  , respectively. Reprinted with permission from [13]. Copyright 2015 American Chemical Society.
, respectively. Reprinted with permission from [13]. Copyright 2015 American Chemical Society.
Download figure:
Standard image High-resolution image5.4. Comparison of intramolecular MD force fields
We have carried out a comparison of the three reactive potentials used to describe intramolecular bonds covered in this review—the Tersoff, REBO, and AIREBO potentials. Here we compare the effect that these three different models have on the displacement energy for removing individual C and H atoms from coronene. To determine the displacement energies independent of projectile type, we perform the simulations without using a projectile. Instead we displace a single atom (the PKA) along a predetermined initial trajectory. The lowest kinetic energy required to promptly remove that atom from the molecule is then taken to be the displacement energy for that specific initial path. The displacement energy of a given atom in coronene is determined along 500 evenly distributed trajectories.
Due to its symmetry, there are only four unique atom positions of the 36 atoms in coronene—three for C and one for H (see figure 18). The displacement energies of each position were determined over all angles and used to calculate distributions of the displacement energy for the entire coronene molecule. The simulations were performed using the LAMMPS MD package [115, 116] and the Tersoff, REBO, and AIREBO potentials as defined in the software. Parameters for describing the C–C, C–H, and H–H interactions using the Tersoff potential are from [42].

Figure 18. Displacement energies for three unique carbon-, and one unique hydrogen-, positions in coronene as calculated with the Tersoff potential. The calculated displacement energy for a given direction is displayed both by the size and color of the corresponding point: large red points show directions with high displacement energies and small blue points show those with low displacement energies.
Download figure:
Standard image High-resolution imageFigure 18 shows the angular distributions of the displacement energy for the four unique atom positions in coronene calculated with the Tersoff potential. The color and size of each point in the figure represents the displacement energy of the enclosed atom along that trajectory. The highest displacement energies are found for trajectories that follow the bonds to the nearest neighboring atoms and can surpass 100 eV in a few extreme cases. For the two positions where a C atom is bound to three other C atoms, the lowest displacement energies are obtained along trajectories perpendicular to the molecular plane. With the Tersoff potential the value for these is on average about 22 eV. The lowest displacement energies for removing a C atom is found for the peripheral atoms that are bound to an H atom, which can be as low as 17 eV along trajectories close to parallel to, but away from, the molecular disk. The displacement energy for knocking out an H atom is more uniform in its angular distribution at about 9 eV. The values of the displacement energy differ between the different force fields, but their angular dependent behavior remains largely the same as shown in figure 18 and agree well with results from simulations of graphene [117].
Total displacement energy distributions calculated with each of the three force fields for all atoms are shown in figure 19. Here the total distributions over all displacement angles are shown in red and the contribution from angles, θ, less than 60 degrees from perpendicular to the molecular plane are shown in blue. All three total distributions show a strong peak at energies around 25–30 eV which is mainly the result of knockout trajectories out of the molecular plane (in blue) and from peripheral C atoms that have low displacement energies away from the molecular disk (see figure 18). The Tersoff and REBO potentials give very similar results, peaking around 27 eV with the AIREBO potential giving on average somewhat higher displacement energies as well as a more pronounced tail towards high energies.
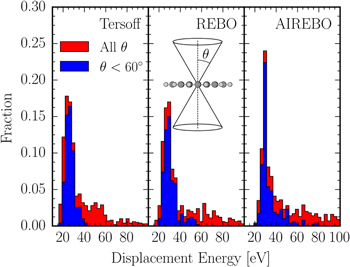
Figure 19. Distributions of displacement energies for prompt knockout of a single C atom from coronene using classical MD simulations with, from left to right: the Tersoff, REBO, and AIREBO force fields. The red distributions cover all angles, θ, and the blue distributions only trajectories where the atom is displaced at an angle  (see inset of middle panel).
(see inset of middle panel).
Download figure:
Standard image High-resolution imageThe mean displacement energies for the removal of a carbon atom along any initial trajectory (red distributions in figure 19) are 35.4 eV, 45.1 eV, and 48.3 eV for the Tersoff, REBO, AIREBO potentials, respectively. The corresponding values for trajectories out of the molecular plane ( , blue distributions in figure 19) are 26.1 eV, 30.2 eV, and 33.2 eV. These out of plane values are in good agreement with the displacement energies predicted by Postma et al for trajectories perpendicular to the molecular plane (
, blue distributions in figure 19) are 26.1 eV, 30.2 eV, and 33.2 eV. These out of plane values are in good agreement with the displacement energies predicted by Postma et al for trajectories perpendicular to the molecular plane ( ) when using the REBO potential [12].
) when using the REBO potential [12].
6. Bond-forming reactions
6.1. Increased reactivity
Intact fullerenes and PAH molecules are stable and not particularly reactive molecular systems. However, when an atom is promptly removed from one of these molecules the result is often a highly reactive fragment. Figure 20 shows an example of a circumcoronene molecule where a single C atom has been removed from the innermost hexagonal ring. The left panel in this figure shows the structure of this relaxed C53H18 fragment seen face-on. Around the defect caused by the removal of a C atom we see that there are three C atoms left with dangling bonds, two identical atoms labelled as 1 and a single atom labelled as 2. All three of these sites are highly reactive and the right panel in figure 20 shows the structure of the system when a phenyl group (C6H5) forms bonds with one of the atoms in the fragment at position 1 [82]. The binding energy of the phenyl radical to this position is about 4 eV for a neutral system and 4.4 eV if the combined C59H23 system is positively charged [82]. These binding energies are significantly higher than when the phenyl group is bound to an intact circumcoronene molecule at the same position, at 0.2 eV and 0.9 eV for the neutral and cation systems, respectively [82]. Chen et al found similar trends for several reaction partners—the reactivity of PAH molecules is significantly increased when an atom is removed by knockout [82]. This is consistent with results from a theoretical study on the role of defects in the reactivity of graphene [11].
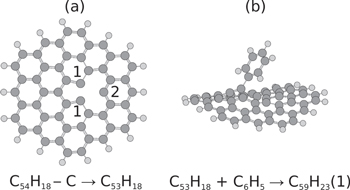
Figure 20. (a) Relaxed structure of circumcoronene (C54H18) when a single C atom has been removed from the innermost ring. This structure has three C atoms with unpaired bond sites, two identical positions labelled 1, and a single one labelled 2. (b) A phenyl radical has formed a covalent bond at position number 1. This bond is about a factor of 20 times stronger than it would be for an intact PAH molecule. Reprinted with permission from [82]. Copyright 2014, AIP Publishing LLC.
Download figure:
Standard image High-resolution image6.2. Reactions in clusters
When non-statistical fragmentation takes place inside of molecular clusters, newly formed radicals may react with neighboring molecules and efficient molecular growth can take place. Delaunay et al detected widespread molecular growth in experiments at the ARIBE facility where ions collided with PAH clusters [58]. Starting with loosely bound clusters of pyrene (C16H10) they detected covalently bound molecules containing up to more than 30 C atoms formed in collisions with ions at center-of-mass energies above 10 keV. For light projectiles (e.g. He) this energy range is well into the regime where electronic stopping dominates, but by varying the mass and velocity of the projectiles they concluded that nuclear scattering processes were responsible for the observed growth [58]. Here, the fast redistribution of the excess energy among all cluster constituents is crucial as it allows the molecular growth products to be formed sufficiently cold to survive on the experimental timescale (see section 4.2).
The left panels of figure 21 shows experimental mass spectra measured by Delaunay et al for collisions between pyrene clusters and 11 keV He+ ions (bottom row) and 12 keV Ar2+ ions (top row) [58]. In both mass spectra we see the range of masses that include the intact molecular monomer (C16 ) and loosely bound dimers ([C16H10]
) and loosely bound dimers ([C16H10] ) that remain after the collisions. However, with 12 keV Ar2+ projectiles we also see a wide range of peaks coming from products with more than 16 C atoms. These features were not unique to the collisions with Ar2+, but decreased in intensity with lighter and faster projectiles. With the lightest projectiles, like He+, these features are mostly absent [58].
) that remain after the collisions. However, with 12 keV Ar2+ projectiles we also see a wide range of peaks coming from products with more than 16 C atoms. These features were not unique to the collisions with Ar2+, but decreased in intensity with lighter and faster projectiles. With the lightest projectiles, like He+, these features are mostly absent [58].

Figure 21. Experimental and simulated mass spectra for Ar (top row) and He (bottom row) ions/atoms colliding with clusters of pyrene molecules at 12 and 11 keV, respectively [58]. The left panels show the experimental mass spectra. With Ar2+ projectiles a wide range of larger molecules containing between 17 and 37 C atoms are detected, but with He projectiles these are mostly absent. The right panels are mass spectra from classical MD simulations of the same collisions. The simulated data are shown with an artificial gaussian smoothing with widths of 1 amu to approximately match the resolution of the experiment. The simulations reproduce the details regarding molecular growth seen in the experiments. The inset in the lower right panel shows a schematic of a collision and some of the new molecules that are observed in the simulations [58].
Download figure:
Standard image High-resolution imageIn connection to the experiments, Delaunay et al also performed classical MD simulations of the collisions and bond-forming reactions [58]. In that work the entire collision processes between keV ions and the PAH clusters—and the following bond-forming reactions—could be followed in simulations extending over picosecond timescales. Using the AIREBO potential to describe the molecular bonds and dispersion forces in the cluster, and the ZBL potential for modeling the collision interactions, these simulations were able to reproduce the detailed features in the experimental mass spectra as seen in the right panels of figure 21. SCC-DFTB simulations were then used to test the accuracy of the classical MD simulations with regards to bond formation and the effect of charge on the molecules formed. The results showed that the classical MD simulations gave structures more rigid than those found with quantum chemical methods (regarding preservation of bond angles). However, the reactivity of the fragments and the ability to form larger molecules was confirmed by the SCC-DFTB simulations [58].
Similar bond-forming reactions have also been detected in clusters of fullerene molecules. In a study of 22.5 keV He2+ ions colliding with C60 clusters, Zettergren et al detected the formation of covalently bound dumbbell shaped  and
and  molecules [52, 57]. These were produced when the ion knocked out one or two atoms from molecules within the cluster. The remaining
molecules [52, 57]. These were produced when the ion knocked out one or two atoms from molecules within the cluster. The remaining  or
or  fragments could then react with neighboring molecules. Coincidence measurements showed that the
fragments could then react with neighboring molecules. Coincidence measurements showed that the  and
and  products were strongly correlated with the detection of intact
products were strongly correlated with the detection of intact  ions, the result of penetrating collisions that lead to multiple ionization of the cluster. Zettergren et al also used classical MD simulations with the Tersoff potential to study the reactions between
ions, the result of penetrating collisions that lead to multiple ionization of the cluster. Zettergren et al also used classical MD simulations with the Tersoff potential to study the reactions between  fragments and intact C60 that lead to the formation of
fragments and intact C60 that lead to the formation of  [57]. A sequence from simulations of dumbbell
[57]. A sequence from simulations of dumbbell  forming in a cluster at different times is shown in figure 22. These simulations revealed that the barrier for reactions was 1 eV or less [57], almost two orders of magnitude lower than that for forming bonds between two intact C60 molecules [118].
forming in a cluster at different times is shown in figure 22. These simulations revealed that the barrier for reactions was 1 eV or less [57], almost two orders of magnitude lower than that for forming bonds between two intact C60 molecules [118].
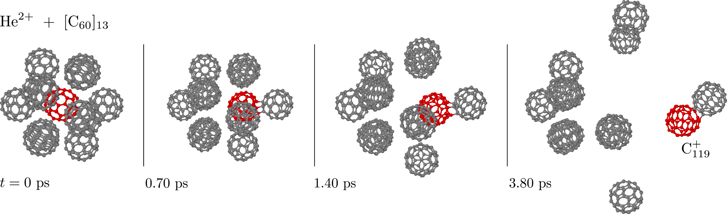
Figure 22. Snapshots at different time steps from a classical MD simulation of  forming in a [C60]13 cluster after the knockout of a single C atom from the central molecule [57]. The
forming in a [C60]13 cluster after the knockout of a single C atom from the central molecule [57]. The  fragment formed at the beginning of the simulation (t = 0 ps) is highlighted in red. Reprinted figure with permission from [57]. Copyright 2013 by the American Physical Society.
fragment formed at the beginning of the simulation (t = 0 ps) is highlighted in red. Reprinted figure with permission from [57]. Copyright 2013 by the American Physical Society.
Download figure:
Standard image High-resolution imageIn light of the high reaction barrier for forming covalently bound  , it is surprising that Zettergren et al detected a strong signal for ions with this mass in the same type of penetrating collisions as dumbbell
, it is surprising that Zettergren et al detected a strong signal for ions with this mass in the same type of penetrating collisions as dumbbell  and
and  [57]. A possible explanation is that [C@
[57]. A possible explanation is that [C@![${{\rm{C}}}_{59}{]}^{+}$](https://content.cld.iop.org/journals/0953-4075/49/16/162001/revision1/jpbaa2dc9ieqn86.gif) is formed when a C atom is displaced within a molecule in the collision. This defective fullerene ion, while having the same mass-to-charge ratio as
is formed when a C atom is displaced within a molecule in the collision. This defective fullerene ion, while having the same mass-to-charge ratio as  , could be as reactive as a
, could be as reactive as a  fragment and thus easily form covalent bonds with a neighboring molecule. Hence a covalently bound
fragment and thus easily form covalent bonds with a neighboring molecule. Hence a covalently bound  , or [C@
, or [C@![${{\rm{C}}}_{119}{]}^{+}$](https://content.cld.iop.org/journals/0953-4075/49/16/162001/revision1/jpbaa2dc9ieqn90.gif) , molecule might also form with a low reaction barrier in the same way as
, molecule might also form with a low reaction barrier in the same way as  and
and  .
.
In a followup study, Yang et al used SCC-DFTB MD simulations and DFT calculations to study the formation of dumbbell  and
and  [89]. Yang et al used these quantum chemical methods to better describe the formation of dumbbell shaped fullerenes and showed that SCC-DFTB produced results in good agreement with more advanced DFT structure calculations at the B3LYP/6-31G(d) level of theory (for these types of systems) as seen in figure 23 [89].
[89]. Yang et al used these quantum chemical methods to better describe the formation of dumbbell shaped fullerenes and showed that SCC-DFTB produced results in good agreement with more advanced DFT structure calculations at the B3LYP/6-31G(d) level of theory (for these types of systems) as seen in figure 23 [89].

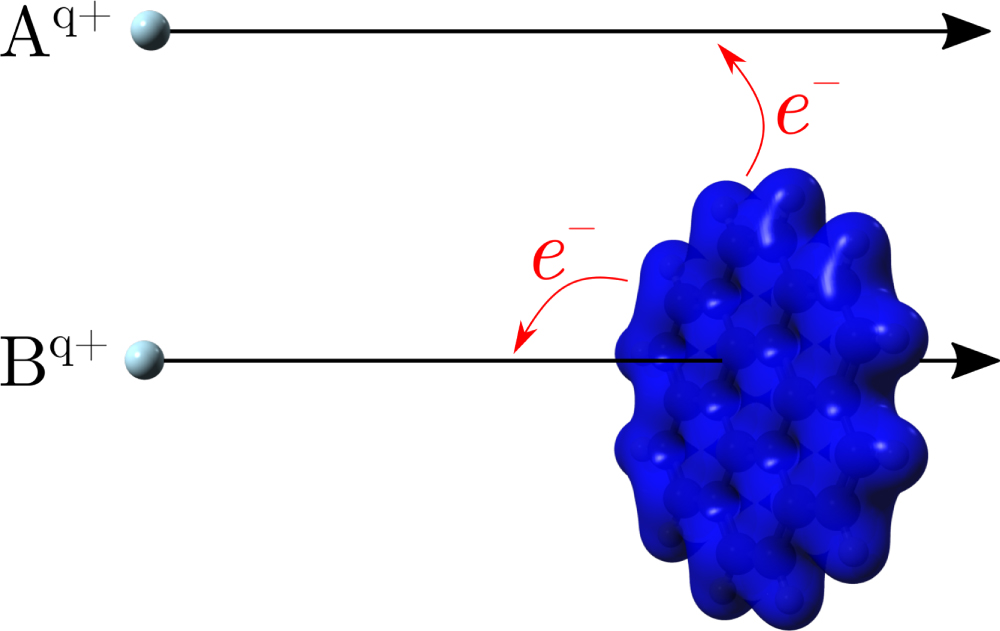{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 23. Potential energy as a function of simulation time for the remaining  fragment after the knockout of one atom from
fragment after the knockout of one atom from  calculated by Yang et al using SCC-DFTB (see text) [89]. The yellow circles show the energies of the same structures calculated at B3LYP/6-31G(d) level of theory [120]. The SCC-DFTB model of
calculated by Yang et al using SCC-DFTB (see text) [89]. The yellow circles show the energies of the same structures calculated at B3LYP/6-31G(d) level of theory [120]. The SCC-DFTB model of  gives accuracies comparable to those of the more advanced DFT method. Reprinted figure with permission from [89]. Copyright 2014 by the American Physical Society.
gives accuracies comparable to those of the more advanced DFT method. Reprinted figure with permission from [89]. Copyright 2014 by the American Physical Society.
Download figure:
Standard image High-resolution image{kind=link}
Dumbbell  and
and  molecules were also detected by Gatchell et al in collisions between 22.5 keV alpha particles and mixed cluster of C60 and coronene [55]. However, they did not detect any bond-forming reactions between C60 and coronene, or between two coronene molecules/fragments. They used the Tersoff potential in MD simulations to study the reactivity of C59 and C23H12 fragments (of coronene) in interactions with intact C60 and intact coronene molecules. These simulations showed that the mixed coronene-fullerene and coronene–coronene reactions had significantly higher reaction barriers than fullerene–fullerene when only a single atom is removed from the system [55].
molecules were also detected by Gatchell et al in collisions between 22.5 keV alpha particles and mixed cluster of C60 and coronene [55]. However, they did not detect any bond-forming reactions between C60 and coronene, or between two coronene molecules/fragments. They used the Tersoff potential in MD simulations to study the reactivity of C59 and C23H12 fragments (of coronene) in interactions with intact C60 and intact coronene molecules. These simulations showed that the mixed coronene-fullerene and coronene–coronene reactions had significantly higher reaction barriers than fullerene–fullerene when only a single atom is removed from the system [55].
The reactions that lead to dumbbell shaped fullerenes are of a very different type than the growth processes previously observed when 200 keV Xe20+ ions impacted very large fullerene clusters [119]. There the collisions led to very strong local heating along the path of the projectile forming a plasma of C atoms and small fragments. The plasma then began to cool and to self-assemble or coalesce with neighboring molecules resulting in a broad distribution of larger fullerenes [119]. The competition between these growth processes and the statistical decay of hot reaction products led to both bottom-up and top-down fullerene formation processes being observed in these experiments [119].
7. Concluding remarks and outlook
In this topical review we have discussed recent advances in the understanding of ion/atom interactions with isolated PAHs, fullerenes and their clusters. The main focus has been on prompt atom knockout in billiard-ball-like nuclear scattering processes. These studies show that:
(1) Atom knockout is important for collision energies in the energy range from ∼20 eV up to hundreds of keV.
(2) Such non-statistical fragmentation processes may lead to unique and highly reactive fragments which are not formed in statistical fragmentation processes.
(3) Direct evidence for knockout has been identified for isolated PAHs and fullerenes. This is possible due to their unique mass spectrometric fingerprints (single carbon loss) and large heat capacities that limit the influence from secondary fragmentation processes.
(4) Efficient molecular growth processes have been observed in clusters of PAHs and fullerenes. Here, the excitation energy is rapidly shared by all the molecules in a cluster making the colliding molecule much colder than if it would have been isolated. This allows the reactive knockout fragments to form covalent bonds with neighboring molecules in the cluster before it disintegrates on picosecond timescales. The so formed growth products are sufficiently cold to survive on the experimental microsecond timescales.
(5) Knockout driven reactions are expected to be important for any complex molecular system, but are in general hard to identify due to the lack of clear fingerprint signatures and due to strong secondary fragmentation processes. However, this may be possible by embedding them in clusters (see point 4). Future experimental and theoretical studies will explore the consequences of atom knockout in e.g. biomolecules, and the role of a surrounding (cluster) environment.
Effects of ion irradiation have been studied rather extensively with nano-structured materials [6] and, there, material modifications resulting from the displacements of individual atoms have been discussed intensively. Much less has so far been done on the level of single molecules. For this purpose, more controlled studies of charge transport, energy flow, and molecular growth processes in clusters are needed. This may help to answer fundamental questions such as 'Are aromatic molecules formed by ion impact on clusters of small non-aromatic hydrocarbon molecules?' and 'Are also fullerenes formed under such conditions?'. Additional examples of future intriguing studies may include measurements of displacement and dissociation energies, and of lifetimes and cooling rates of knockout fragments using e.g. electrostatic storage devices [34, 121–125].
Parallel developments of new theoretical tools will be essential for interpreting the experimental results, for instance quantum chemical approaches treating the correlation between hard nuclear collisions and inelastic losses due to electronic excitations in the collisions [111]. Such joint experimental and theoretical efforts are key to driving this field forward and may help to gauge the significance of knockout processes in, e.g., various astrophysical environments.
Acknowledgments
This work was supported by the Swedish Research Council (Contract No 621-2012-3660). The authors would like to acknowledge Henrik Cederquist, Henning T Schmidt, and Thomas Schlathölter for their many useful comments on the manuscript and our colleagues at Stockholm University, the research groups in Caen, Madrid, and Groningen, and the COST action CM1204 'XUV/X-ray light and fast ions for ultrafast chemistry (XLIC)'.