
Abstract
The ultrafast decay of highly excited electronic states is resolved with a molecular clock technique, using the vibrational motion associated to the ionic bound states as a time-reference. We demonstrate the validity of the method in the context of autoionization of the hydrogen molecule, where nearly exact full dimensional ab-initio calculations are available. The vibrationally resolved photoionization spectrum provides a time–energy mapping of the autoionization process into the bound states that is used to fully reconstruct the decay in time. A resolution of a fraction of the vibrational period is achieved. Since no assumptions are made on the underlying coupled electron–nuclear dynamics, the reconstruction procedure can be applied to describe the general problem of the decay of highly excited states in other molecular targets.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The measurement of ultrafast electronic processes has become an active field of research in the last decade [1–5]. Such measurements usually require attosecond time resolution. It can be achieved, for example, by using phase-synchronized femtosecond pulses in XUV pump—IR probe setups [6–8] or using attosecond pulses in combination with a well-defined time-resolved physical process [9–12]. In high harmonic spectroscopy, for instance, the time–energy mapping between the harmonic energy and the underlying mechanism of ionization and subsequent recombination is used to perform time-resolved measurements with sub-femtosecond resolution [9, 11, 12]. Equivalent approaches are used in core-hole clock spectroscopy, where the Auger decay time provides the reference [10, 13], or in the attoclock technique, where the time reference is provided by the ionization events due to circularly polarized field [14, 15].
Following these schemes, we here aim to time-resolve the fast electronic decay of highly excited states in the hydrogen molecule using the vibrations of the molecular bond as a time reference. This concept, known as molecular clock, was previously employed to track the dynamics associated to the non-sequential double ionization process of the hydrogen molecule [16]. The method relies on the coherence between the vibrational and electronic phase at the initial stage of the dynamics. This coherence allows one to achieve a time resolution that is just a fraction of the vibrational period of the molecule. We extend the concept of molecular clock to bound vibrational and electronic states and use it to follow in time the decay of a resonant electronic state. Specifically, the concept is demonstrated by time-resolving the autoionization (AI) decay of the Q1 state of the H2 molecule, which has a lifetime of a few fs.
Attempts to extract the lifetimes of resonant states for which the vibrational motion plays a significant role were performed in the past using a variety of approaches [13, 17–20]. In contrast to the latter cases, in AI of H2 (and D2), multiple ionization channels interfere, significantly modifying the photoelectron spectra. The interference resulting after molecular autoionization was experimentally demonstrated already in the nineties in H2 (and D2) photoionization by synchrotron radiation, where its signature was captured in the energy differential cross sections [21–23], as well as in the molecular frame photoelectron angular distributions [24–26]. Along with those works, the first full dimensional ab initio calculation including nuclear and electronic degrees of freedom was also reported [27–30], confirming the essential role of nuclear motion to describe these interferences, as proposed in existing theoretical works using semiclassical approaches in the dissociative ionization channel [31–33]. In the present work, we aim to demonstrate how to extract the dynamical information that is encoded in the non-dissociative ionization probabilities, by applying a reconstruction procedure that accurately reproduces the result of the ab initio calculations.
The proposed method can be extended to other molecules containing light atoms provided that state-resolved vibrational progressions that involve these light atoms can be resolved spectroscopically. The method could be applied whenever the interference between competing ionization pathways is important and is not limited to doubly excited states but could also be applied to Auger decay processes.
1.1. The concept of 'molecular clock'
Ionization of the H2 molecule after absorption of a photon with an energy of ∼30 eV proceeds via two paths (see figure 1). The first ('direct') channel corresponds to leaving a vibrationally excited  ion in the ground electronic
ion in the ground electronic  state, starting the 'molecular clock'. The second ('resonant') channel corresponds to populating the Q1 series of doubly-excited states accesible by one-photon absorption. These states spontaneously decay into the electronic continuum associated to the ground electronic state of
state, starting the 'molecular clock'. The second ('resonant') channel corresponds to populating the Q1 series of doubly-excited states accesible by one-photon absorption. These states spontaneously decay into the electronic continuum associated to the ground electronic state of  by emitting a photoelectron, through the process of AI. The interference of the 'direct' and the 'resonant' ionization channels records their relative phases by mapping them into amplitude modulations of the population of vibrational state in the
by emitting a photoelectron, through the process of AI. The interference of the 'direct' and the 'resonant' ionization channels records their relative phases by mapping them into amplitude modulations of the population of vibrational state in the  electronic ground state as a function of the absorbed energy [22, 23, 27, 34].
electronic ground state as a function of the absorbed energy [22, 23, 27, 34].
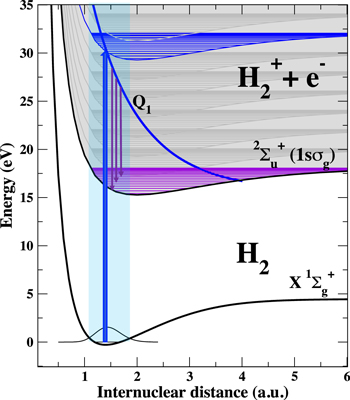
Figure 1. A sketch of the decay of the autoionizing Q1 state of H2 molecule after excitation by a 30 eV photon. The photon absorption simultaneously populates the background electronic continuum associated to the ground state of the ion, 1s , depicted by a 'discretized' continuum. At any electron energy we include the vibrational structure for the bound ionic states. The excitation creates a vibrational wavepacket on the Q1 state potential that starts accelerating and moving towards higher internuclear distances. As the Q1 state undergoes an AI decay by emitting a photoelectron, the vibrational wavepacket decays into bound vibrational states of the
, depicted by a 'discretized' continuum. At any electron energy we include the vibrational structure for the bound ionic states. The excitation creates a vibrational wavepacket on the Q1 state potential that starts accelerating and moving towards higher internuclear distances. As the Q1 state undergoes an AI decay by emitting a photoelectron, the vibrational wavepacket decays into bound vibrational states of the  ground state. Due to the momentum conservation, higher final vibrational states are populated at later decay times.
ground state. Due to the momentum conservation, higher final vibrational states are populated at later decay times.
Download figure:
Standard image High-resolution imageContinuum resonances—states that decay by AI—are present in all multielectronic atoms and molecules. In atoms, they lead to the well known Fano resonances [35]. In molecules, the picture can be largely modified by the coupling between electronic and vibrational degrees of freedom [36, 37], resulting even in the dissapearance of the Fano-like profiles in the total photoionization yield. An extreme case is the H2 molecule. The lifetime of its lowest resonant state, that is also most largely populated by one-photon absorption, is of the order of a few fs (1–3 fs). However, the light mass of the nuclei confers a rapid vibrational dynamics on a comparable time-scale. Consequently, in the H2 molecule, the ultrafast electronic decay is strongly coupled to the vibrational dynamics of the molecule [27, 38]. A reconstruction procedure that retrieves both phase and amplitude of the 'resonant' wave packet would yield complete information about its corresponding time evolution. Such reconstruction is possible via a time–energy mapping between the AI decay and the vibrational dynamics in the final  ion, which is illustrated in figure 1. The decay from the Q1 band of autoionizing states populates different vibrational levels of the
ion, which is illustrated in figure 1. The decay from the Q1 band of autoionizing states populates different vibrational levels of the  ion at different time-delays between excitation (XUV pump) and autoionization (probe) [38]. The longer the pump-probe delay, the higher is the energy of the populated vibrational states. The time–energy mapping is similar to the one used in the attosecond streak-camera [3, 39] although here the streaking is due to the accelerating motion of the nuclei.
ion at different time-delays between excitation (XUV pump) and autoionization (probe) [38]. The longer the pump-probe delay, the higher is the energy of the populated vibrational states. The time–energy mapping is similar to the one used in the attosecond streak-camera [3, 39] although here the streaking is due to the accelerating motion of the nuclei.
In the following, we demonstrate how the AI decay can be fully reconstructed in time from the vibrationally resolved photoionization spectra of the H2 molecule, if the 'direct' ionization channel is known or, at least, can be approximated from the experimental data. This reconstruction renders a simple qualitative picture of the time–energy mapping, where each final vibrational state ν provides a temporal attosecond gate for the AI decay. In order to validate the reconstruction procedure, we use the simulated data produced by nearly-exact calculations whose reliability has been amply verified in previous works by comparing them with experimental data [40]. Moreover, the reconstruction procedure presented in this work is general and does not rely on any assumptions or approximations about the underlying dynamics, which are usually performed en route to calculate the dynamics of the 'resonant' states in molecules.
2. Methodology
2.1. Photoionization spectra
In the basic formalism of one-photon ionization close to a resonant state, we consider a molecule in its ground state  that is excited by a laser field to a manifold of excited states
that is excited by a laser field to a manifold of excited states  through the interaction potential V(t). We assume that
through the interaction potential V(t). We assume that  is orthogonal to
is orthogonal to  by construction. Within the first order of perturbation theory, the excited wavefunction
by construction. Within the first order of perturbation theory, the excited wavefunction  is (atomic units are used throughout)
is (atomic units are used throughout)
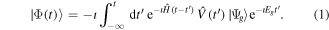
Consider an observable that projects  onto the final state
onto the final state  , which is stationary, non-decaying and has energy
, which is stationary, non-decaying and has energy  . We can further define a state vector
. We can further define a state vector  that contains all the remaining states of the system, such that
that contains all the remaining states of the system, such that  . Thus, we divide the system into a subspace that includes the final stationary state
. Thus, we divide the system into a subspace that includes the final stationary state  , and a subspace that includes all the intermediate states
, and a subspace that includes all the intermediate states  . These states include resonances that are assumed to irreversibly decay into
. These states include resonances that are assumed to irreversibly decay into  as
as  . Hence, the dynamics in each subspace is non-hermitian. The interaction between the two subspaces is governed by
. Hence, the dynamics in each subspace is non-hermitian. The interaction between the two subspaces is governed by  , whose contribution to
, whose contribution to  is assumed to vanish as
is assumed to vanish as  , allowing to define
, allowing to define  as the final state after ionization. The outcome of such measurement is
as the final state after ionization. The outcome of such measurement is
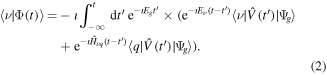
The approach could be generalized to more complicated cases, although the reconstruction would be more involved. In the case of two distinct intermediate subspaces  and
and  , it would be enough to separate the
, it would be enough to separate the  and
and  from the
from the  manifold. Defining the laser field
manifold. Defining the laser field  via its Fourier transformation
via its Fourier transformation  , where
, where  is the spectral density of the pulse and
is the spectral density of the pulse and  is the dipole operator and introducing the new time variable
is the dipole operator and introducing the new time variable  the expression (2) above becomes
the expression (2) above becomes
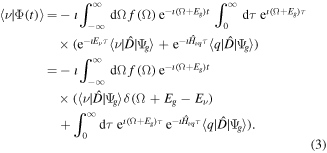
The resulting amplitude is thus described by a sum of two terms—a 'direct' contribution (first term in the brackets), which is the 1st order interaction with the laser, and a 'resonant' contribution (second term in the brackets), which is mediated by the non-stationary dynamics of the system, e.g. decay of resonances and transient states. These two terms determine the density of probability  of an energy-resolved measurement, e.g., the energy resolved spectra:
of an energy-resolved measurement, e.g., the energy resolved spectra:
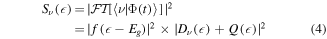
with
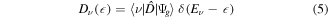
and
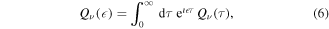
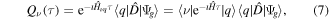
where  denotes the Fourier transformation.
denotes the Fourier transformation.
2.2. Reconstruction of the 'resonant' ionization path
The full spectrum  is the result of the interference between the 'direct' and the 'resonant' ionization paths. Hence, if both the spectrum
is the result of the interference between the 'direct' and the 'resonant' ionization paths. Hence, if both the spectrum  and the 'direct' ionization paths are measurable or predictable, e.g., from a theoretical calculation, the 'resonant' contribution can be obtained using a procedure similar to heterodyne detection. In the case of single photoionization of H2, and following equation (4), the full vibrationally resolved photoionization spectrum
and the 'direct' ionization paths are measurable or predictable, e.g., from a theoretical calculation, the 'resonant' contribution can be obtained using a procedure similar to heterodyne detection. In the case of single photoionization of H2, and following equation (4), the full vibrationally resolved photoionization spectrum  can be expressed as
can be expressed as
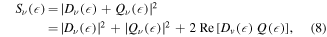
where the corresponding ionization dipole elements are  for the 'direct' and
for the 'direct' and  for the'resonant' paths,
for the'resonant' paths,  is the final photoelectron energy and ν is the final vibrational state label. Here and in the following, a monochromatic laser pulse is considered, for which
is the final photoelectron energy and ν is the final vibrational state label. Here and in the following, a monochromatic laser pulse is considered, for which  with
with  . Nevertheless, all the results can be straightforwardly generalized to pulses of finite duration by using the spectral density of the pulse
. Nevertheless, all the results can be straightforwardly generalized to pulses of finite duration by using the spectral density of the pulse  , for example
, for example
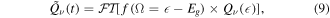
which is exact for single photon processes. Also, pulses of finite duration will be considered in section 3.3. We further assume that  , i.e., that the 'resonant' ionization channel is weaker than the 'direct' channel. This condition is usually satisfied in the case of doubly excited states, and is also satisfied in the case of H2 ionization considered here—the two-electron excitation upon one-photon absorption is coming purely from the electron correlation terms and therefore are appreciably smaller than the promotion of an electron into the continuum associated to the ground state of the cation. In practice, the reconstruction procedure outlined below works well even when the strength of the 'direct' and 'resonant' channels are comparable. Furthermore,
, i.e., that the 'resonant' ionization channel is weaker than the 'direct' channel. This condition is usually satisfied in the case of doubly excited states, and is also satisfied in the case of H2 ionization considered here—the two-electron excitation upon one-photon absorption is coming purely from the electron correlation terms and therefore are appreciably smaller than the promotion of an electron into the continuum associated to the ground state of the cation. In practice, the reconstruction procedure outlined below works well even when the strength of the 'direct' and 'resonant' channels are comparable. Furthermore, ![$\mathrm{Im}[{D}_{\nu }(\epsilon )]\approx 0$](https://content.cld.iop.org/journals/0953-4075/50/14/144001/revision2/jpbaa7215ieqn48.gif) is assumed, i.e., that the scattering phase of 'direct' photoionization is negligible. Indeed, the scattering phase is large when multi-electron/multi-channel effects are involved, and small for single-electron ionization, described by the
is assumed, i.e., that the scattering phase of 'direct' photoionization is negligible. Indeed, the scattering phase is large when multi-electron/multi-channel effects are involved, and small for single-electron ionization, described by the  ionization dipole. Using the assumptions above, the terms in equation (8) can be rearranged to obtain an explicit expression for the 'resonant' ionization path:
ionization dipole. Using the assumptions above, the terms in equation (8) can be rearranged to obtain an explicit expression for the 'resonant' ionization path:
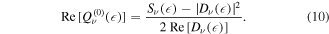
Although equation (10) allows one to obtain only the real part of the 'resonant' term, it still contains the full information about the decay. This is the case, because in the equation (6) the time integral is performed from 0 to  and not from
and not from  to
to  . Thus, the full time-dependent AI decay
. Thus, the full time-dependent AI decay  into the final state ν can be recovered by performing a Fourier transformation, as is evident from equation (6). We will demonstrate next that the quantity
into the final state ν can be recovered by performing a Fourier transformation, as is evident from equation (6). We will demonstrate next that the quantity  that is recovered corresponds to the rate of population transfer from the state Q1 to state ν.
that is recovered corresponds to the rate of population transfer from the state Q1 to state ν.
Since the  term was neglected in equation (8), the reconstructed 'resonant' contribution
term was neglected in equation (8), the reconstructed 'resonant' contribution  from equation (10) is only approximate. It can be used to estimate the approximate spectra
from equation (10) is only approximate. It can be used to estimate the approximate spectra  . By inserting the difference
. By inserting the difference  into equation (10), instead of
into equation (10), instead of  , a correction to the 'resonant' term
, a correction to the 'resonant' term  is obtained. It can then be used to estimate a higher order
is obtained. It can then be used to estimate a higher order  . Recursively repeating this procedure, an increasingly more accurate solution
. Recursively repeating this procedure, an increasingly more accurate solution  is obtained. We found accurate results for the 3rd order solution.
is obtained. We found accurate results for the 3rd order solution.
3. Results and discussion
3.1. Vibrationally resolved photoionization spectra
The vibrationally resolved photoionization spectra of H2 are presented in figure 2. The spectra were obtained by solving the full dimensional time-dependent Schrödinger equation (TDSE) [40] and employing a 400 as long XUV pulse with frequency centered around 30 eV and a laser intensity of 109 W cm−2. For the laser field parameters used here, photoionization takes place within the perturbative regime, where we can define a cross section that is independent of the pulse parameters (length, intensity, etc). The spectra shown in figure 2 are the normalized probabilities to the spectral intensity of the laser pulse as described in [41] to obtain the laser-field independent ionization probability, i.e. and effective vibrationally resolved photoionization cross section. Although an attosecond pulse was used in the calculation, an analogous spectra would be obtained using a time-independent calculation as in previous works asuming monochromatic radiation [27]. In an experiment, such correlated spectra could be obtained by, e.g., measuring the photoelectrons and  fragments in coincidence after ionization by an attosecond pulse [42]. However, the present scenario would require to additionally measure the vibrational state of the
fragments in coincidence after ionization by an attosecond pulse [42]. However, the present scenario would require to additionally measure the vibrational state of the  . Alternatively, analogous spectra could be obtained by using a monochromatic laser beam and measuring the photoionization spectra at different laser frequencies, as it was done in [43].
. Alternatively, analogous spectra could be obtained by using a monochromatic laser beam and measuring the photoionization spectra at different laser frequencies, as it was done in [43].
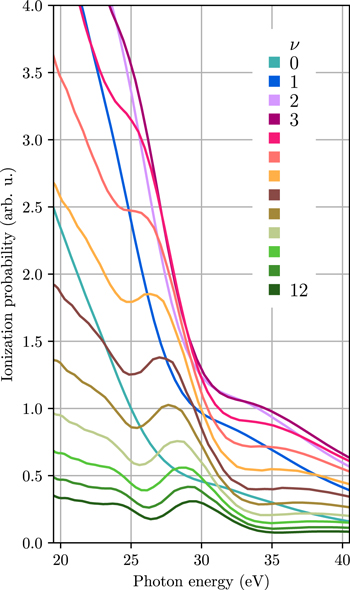
Figure 2. Vibrationally resolved photoionization spectra for the lowest  final vibrational states.
final vibrational states.
Download figure:
Standard image High-resolution imageThe methodology employed to obtain a numerical solution of the TDSE for the H2 molecule is described in detail in [40, 44–46] and will not be presented here. In short, it uses a full expansion of the wavefunction into products of electronic and vibrational Born–Oppenheimer states of H2. It allows to obtain 'full' spectrum including the effects of vibrational motion and the AI decay. Additionally, the method allows one to selectively truncate the simulation to a given number of desired states or to suppress the coupling between selected eigenstates. It is thus straightforward to remove the Q1 state manifold such that one obtains the spectrum of a structureless 'direct' photoionization process, i.e. the incoherent background of the 'direct' ionization channel without the 'resonant' contribution. This calculation was used as the 'reference' photoionization channel in the reconstruction. In the abscence of accurate calculation, it could be also approximately extracted from an experimental measurement by extrapolating the data in the energy regions where only direct photoionization is expected.
The shape of vibrationally resolved photoionization spectra can be explained in terms of semi-classical dynamics. It was previously studied in [37] using semi-classical methods for the dissociative ionization channel, with experimental realization in [47]. More recently, it was extended to the photoionization process associated to the bound states of the ion [38]. There, it was shown that the modulations of the photoionization spectra result from the phase accumulated by vibrational wavepacket moving on the Q1 electronic surface, which is in turn directly related to its classical momentum. Once the vibrational wavepacket is created on the Q1 electronic surface it starts gaining momenta and therefore preferentially decays into higher ν vibrational states that are characterized by a higher momentum value. Hence, higher ν states are populated at larger delayed times.
3.2. Reconstruction of the AI decay
We illustrate the reconstruction procedure selecting three vibrational states ν = 0, 6 and 12 that represent low, middle and high energy states from the vibrational progression in the  ground state, although it works equally well for any final (bound) vibrational states. The 'full' photoionization spectra for the selected states are shown in figure 3, together with the 'direct' spectra, which were obtained by removing the Q1 state from the basis of the ab initio calculation. By following the recursive procedure described in the previuos section, we obtain the time-dependent decay rates
ground state, although it works equally well for any final (bound) vibrational states. The 'full' photoionization spectra for the selected states are shown in figure 3, together with the 'direct' spectra, which were obtained by removing the Q1 state from the basis of the ab initio calculation. By following the recursive procedure described in the previuos section, we obtain the time-dependent decay rates  . The quantity
. The quantity  , as defined in equation (6) describing the 'resonant' ionization channel, thus corresponds to the rate of population transfer from the Q1 state to each of the
, as defined in equation (6) describing the 'resonant' ionization channel, thus corresponds to the rate of population transfer from the Q1 state to each of the  final states. These rates are plotted as a function of time in figure 4 for the ν = 0, 6 and 12 final vibrational states. The reconstructed AI decay rates reveal that each final vibrational state ν is dominantly populated at a different time
final states. These rates are plotted as a function of time in figure 4 for the ν = 0, 6 and 12 final vibrational states. The reconstructed AI decay rates reveal that each final vibrational state ν is dominantly populated at a different time  after ionization. The larger the vibrational number ν, the longer the delayed time. Therefore, each final vibrational state acts as a temporal gate, capturing a 'time-frame' of the decay of the Q1 doubly excited state. The time resolution achieved between different states ν is just a fraction of a femtosecond.
after ionization. The larger the vibrational number ν, the longer the delayed time. Therefore, each final vibrational state acts as a temporal gate, capturing a 'time-frame' of the decay of the Q1 doubly excited state. The time resolution achieved between different states ν is just a fraction of a femtosecond.
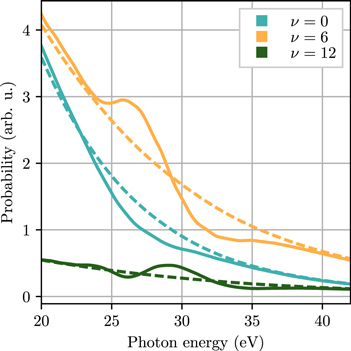
Figure 3. Full (full lines) and direct only (broken lines) vibrationally resolved ionization probabilities for the ν = 0, 6 and 12 vibrational states obtained from ab initio calculations.
Download figure:
Standard image High-resolution image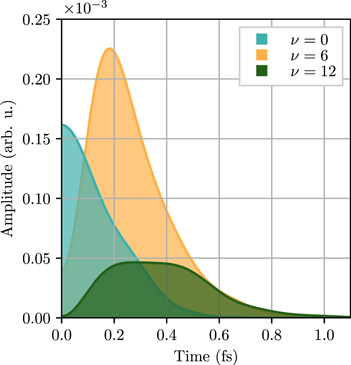
Figure 4. Reconstructed population transfer rates  into ν = 0, 6 and 12 vibrational states as a function of time after ionization.
into ν = 0, 6 and 12 vibrational states as a function of time after ionization.
Download figure:
Standard image High-resolution imageThe integral of the corresponding decay rates  over time yields the total population that decays into each final vibrational state
over time yields the total population that decays into each final vibrational state  . By adding-up these partial populations
. By adding-up these partial populations  , the total population P(t) of the bound part of the
, the total population P(t) of the bound part of the  ground state can be retrieved:
ground state can be retrieved:
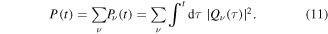
Note, however, that the integral is performed over the modulus of the decay rates  . This is justified, since for a given final energy each final vibrational state constitutes an ionization continuum associated to a unique photoelectron state. Therefore, an incoherent integral over the decay time can be taken to extract the total final population.
. This is justified, since for a given final energy each final vibrational state constitutes an ionization continuum associated to a unique photoelectron state. Therefore, an incoherent integral over the decay time can be taken to extract the total final population.
The accumulated population in the progression of the bound vibrational states ( ) of the
) of the  ground state over time is plotted in figure 5(a), including each individual contribution with a different solid color. The increase of population is entirely due to the decay of the lowest Q1
ground state over time is plotted in figure 5(a), including each individual contribution with a different solid color. The increase of population is entirely due to the decay of the lowest Q1  resonant state and therefore directly reveals the lifetime of this Q1 resonance. The 'direct' ionization of the
resonant state and therefore directly reveals the lifetime of this Q1 resonance. The 'direct' ionization of the  molecule is removed by the reconstruction procedure. The
molecule is removed by the reconstruction procedure. The  ground state is the only open decay channel for the Q1 series of
ground state is the only open decay channel for the Q1 series of  symmetry states, and the lowest state of this series is by far the most dominant autoionization channel.
symmetry states, and the lowest state of this series is by far the most dominant autoionization channel.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. (a) Reconstructed population as a function of time in the bound part of  ground state (the sum over all bound vibrational states
ground state (the sum over all bound vibrational states  ). The contributions of each final state ν to the accumulated population (shaded regions) is indicated; (b) reconstructed partial populations as a function of time for the vibrational states
). The contributions of each final state ν to the accumulated population (shaded regions) is indicated; (b) reconstructed partial populations as a function of time for the vibrational states  and for a 400 as laser pulse (colored lines) compared with the results from an ab initio calculation with the same pulse length (dashed lines).
and for a 400 as laser pulse (colored lines) compared with the results from an ab initio calculation with the same pulse length (dashed lines).
Download figure:
Standard image High-resolution image{kind=link}
3.3. Comparison of reconstructed decay with ab initio calculation
We now compare the reconstruction results with the outcome of the ab initio calculation. Since the dependence on the laser pulse is removed during the reconstruction, the reconstructed dynamics corresponds to an instantaneous excitation of the Q1 state. On the other hand, the ab initio calculation is performed in the time domain, using a finite duration laser pulse. Hence, the finite duration of the laser pulse has to be accounted for in the reconstruction to compare it with the ab initio calculation. This is done by multiplying the reconstructed energy-dependent decay dipoles  by the spectral representation of the laser pulse
by the spectral representation of the laser pulse  . In the time domain this corresponds to a convolution with the laser electric field. The reconstructed decay where the finite pulse duration was taken into account is presented in figure 5(b) (full lines).
. In the time domain this corresponds to a convolution with the laser electric field. The reconstructed decay where the finite pulse duration was taken into account is presented in figure 5(b) (full lines).
In order to obtain the ab initio final state populations due to the resonant ionization path alone, additional truncated ab initio calculations were performed. In these calculations, the non-stationary evolution of Q1 state coupled to the background continua was fully included, but the transition dipole matrix elements between the ground state of H2 and the background continua associated to the ground state of  were set to zero, thus removing the 'direct' ionization channel. In this way, the final vibrational states ν of
were set to zero, thus removing the 'direct' ionization channel. In this way, the final vibrational states ν of  molecule were populated only by the 'resonant' ionization channel via the Q1 state, capturing its time-dependent population decay rate.
molecule were populated only by the 'resonant' ionization channel via the Q1 state, capturing its time-dependent population decay rate.
The population in each final vibrational state ν over time obtained using the truncated ab initio calculation, i.e. removing the contribution of the direct ionization, are plotted in figure 5(b) (dotted lines). Overall, for most of the final vibrational states they show a good agreement with reconstructed populations that account for a finite pulse duration (figure 5(b) full lines). The total population resulting from the extracted rates, equation (11), exhibits an excellent agreement with the results of the ab initio method, thus validating the reconstruction procedure.
4. Discussion and conclusions
We employ a 'molecular clock' method to reconstruct in time the decay of highly excited molecular states from the vibrationally resolved photoionization spectra of H2 molecule. The nuclear motion associated to the bound states of the ion are used as time reference. In particular, applying the principle of heterodyne detection and using the direct ionization spectra as a reference, we have reconstructed phase and amplitude of the ionization contribution due to the Q1 double excited state, a metastable state lying in the electronic continuum.
We show that the reconstructed resonant ionization channel directly corresponds to the rate of population transfer from the Q1 doubly-excited state to the ground state of  ion. Hence, it provides time information on the population of the final vibrational states of the
ion. Hence, it provides time information on the population of the final vibrational states of the  molecule. Combining the time-dependent population of all the final states, allowed us to reconstruct with attosecond resolution the first femtosecond of the AI decay of the Q1 state.
molecule. Combining the time-dependent population of all the final states, allowed us to reconstruct with attosecond resolution the first femtosecond of the AI decay of the Q1 state.
The achieved resolution is sub 1 fs and is not limited by the vibrational period of the molecule, which is possible due to the coherence between electronic and vibrational dynamics at initial times. As a result, the molecular vibrations do not simply lead to the decoherence of the electronic dynamics. Rather, at the initial times, vibrational dynamics provides a coherent time-gate. This gate is just a fraction of a femtosecond and is therefore able to time-resolve the ultrafast AI decay of the Q1 state. The full knowledge of the resonant channel enabled us to obtain the time domain information. This information reveals a time–energy mapping between the decay of the resonant Q1 state and the final vibrational state energy—higher final vibrational states are populated at slightly later times.
Finally, the reconstruction of the AI decay does not depend on the specific parameters of the laser pulse (indeed, the application of equation (10) automatically subtracts the effect of the laser pulse shape). Hence, even if the photoionization spectra are measured with an arbitrary high spectral resolution, e.g., by using synchrotron radiation pulses, this does not affect the time resolution of the reconstruction procedure, as it depends only on the intrinsic molecular dynamics. In this case, the important quantities are the delay between the pump (ionization) and the probe (AI decay), and the acceleration of the vibrational wavepacket on the Q1 state. If both these quantities evolve on comparable timescales, it will be possible to reconstruct the AI decay from the vibrationally resolved photoelectron spectra.
Acknowledgments
We acknowledge the financial support from the FP7 Marie Curie ITN CORINF, the European Research Council under the ERC grant 290853 XCHEM, the European COST Action CM1204 XLIC and the MINECO Project No. FIS2013-42002-R. AP acknowledges Ramón y Cajal Programme from MINECO (Spain). LM acknowledges support from DFG priority programme 1840 QUTIF. Theoretical calculations were obtained at the Mare Nostrum BSC and CCC-UAM computer centers.