
Abstract
The soft matter of biological systems consists of mesoscopic length scale building blocks, composed of a variety of different types of biological molecules. Most single biological molecules are so small that 1 billion would fit on the full-stop at the end of this sentence, but collectively they carry out the vital activities in living cells whose length scale is at least three orders of magnitude greater. Typically, the number of molecules involved in any given cellular process at any one time is relatively small, and so real physiological events may often be dominated by stochastics and fluctuation behaviour at levels comparable to thermal noise, and are generally heterogeneous in nature. This challenging combination of heterogeneity and stochasticity is best investigated experimentally at the level of single molecules, as opposed to more conventional bulk ensemble-average techniques. In recent years, the use of such molecular experimental approaches has become significantly more widespread in research laboratories around the world. In this review we discuss recent experimental approaches in biological physics which can be applied to investigate the living component of soft condensed matter to a precision of a single molecule.
1. Introduction
Single molecule experimentation (SME) is a banner for approaches that measure or manipulate individual molecules that may be in the context of larger systems, for example in the actual complex soft matter of living biological cells, or in highly reduced environments such as single molecule assays in vitro in which perhaps only the one type of purified, isolated single molecule is present. SME has come to fruition over the last 20 years and represents a powerful complement to the predominant approaches using ensemble-averaging. Through measurement of previously unresolved characteristics, the range of new techniques have permitted several familiar molecules to become significantly better understood. These techniques have been applied particularly enthusiastically in biology, and have revealed some profound new aspects of life at the molecular level.
Articles reviewing SME have been published from various perspectives. A recent, broadly encompassing review [1] gives coherent graphical representations of the basic classes of techniques and associated discoveries. In it, the field of SME is decomposed into techniques that primarily use electrons, photons or force to probe or manipulate. Other reviews have focused on the ubiquitous approaches of fluorescence microscopy [2] and force spectroscopy [3]. There are also some extensive reviews written from a biophysics perspective [4, 5], and emphasizing the theoretical character of SME [6].
Our review here assumes that the reader has some awareness of many of the basic experimental techniques that have been ultimately implemented in SME, and looks at SME with respect to what it may achieve conceptually, in terms of the raw information it provides in light of the underlying physical nature of living soft condensed matter. Techniques are presented relating to a progression of abstract measurements or manipulations that one might wish to make and, where appropriate, ensemble-average techniques are referred to. Many techniques could conceivably provide answers to each question, and it is hoped that concurrent presentation of the various approaches may prove useful to the experimentalist. For each case, the physical and chemical specifics of the molecule in question and its surrounding environment should be taken into account since these qualities are ultimately what is of interest, and may predetermine the most suitable technique [7].
In this article SME is presented as a route to discovering how the structure and behaviour of living soft condensed matter can be explained and quantitatively described. The difference between considering living and non-living soft condensed matter is primarily a conceptual one of functionality, involving paradigms for systems level complexes and interaction, such as those of chemical networks. In order that the physico-chemical characteristics of biomolecules can be experimentally determined, the majority of biophysics SME at present is carried out on molecules in vitro, isolated from confounding factors of their native environment. However, there have been recent successes involving SME in the true physiological context of the living cell, and such directions of research are likely to become significantly more widespread in the next few years [8].
This review starts with a discussion of the key qualities of SME in relation to other approaches to the investigation (and in some cases manipulation) of molecules. Some historical context is given, highlighting advances leading to the realization of SME. The following section discusses the relation between single molecules and their environments. The bulk of the article is an exploration of techniques capable of answering the aforementioned focus questions. The questions are loosely ordered by increasing conceptual sophistication of the associated information gained, or manipulation achieved. Finally, section 6 discusses the future of SME including commentary on observed trends and the combination of techniques.
2. Single molecule experimentation
2.1. Historical background
The alternative to single molecule investigation, measuring mean properties of ensembles of molecules, has led to the vast majority of current biochemical knowledge. However, measurement of properties of individual molecules, as opposed to ensembles, enables conceptually new types of information to be obtained. Simply speaking, the mean value for a property of a system produced by a bulk ensemble-average approach may be replaced by a probability distribution of values for that property of the system when a single molecule approach is used. This is made possible by the direct observation of a property of one or many individual molecules. SME enables investigation when the number of molecules of interest is prohibitively small for ensemble measurements. SME can also be seen to subsume the capabilities of ensemble experimentation if the ergodic hypothesis is true for the system under study. This denotes that the average of a measured parameter over a 'long time' (dependent on the system), and the average over the statistical ensemble, should be the same. Regardless of the applicability of the ergodic hypothesis, SME has introduced a brand new set of capabilities.
Heterogeneity, both static and dynamic, is an intrinsic feature of biomolecules and their function. Statically heterogeneous molecules are those that do not play an active role in the interactions of interest of a biological system. In a standard ensemble investigative method, these molecules must be accounted for in analyses in order that their presence can be seen not to have given weight to the results. This step may, however, involve inaccurate approximation. Dynamic heterogeneity refers to inter-conversion of a subgroup of molecules in the system that occurs within the timescale of the observation. This dynamic variation will be lost in the bulk value gained by ensemble study as the stochastic activity of the numerous inter-converting molecules may, in all but some exceptional cases, be unsynchronized. In this case, a single molecule technique may produce one of two types of result that bear further information on the dynamic heterogeneity. If the inter-conversion takes place at a rate that can be effectively sampled by the experimental temporal resolution, the property relating to discrete stages of activity will be discerned. In other words, variation in time of a property may be measured. If the process occurs more rapidly than the technique may resolve, the measured result may represent a weighted time average of the property when the molecule has explored a range of different states. As an example, one might be investigating a molecule's biochemical reaction pathway. The single molecule approach might be to use fluorescence microscopy to record a video of the molecule in question and analyse this to produce a frequency histogram of its dwell times at each state. This may elucidate rare or transient states that might not have been observed with an ensemble method.
SME approaches have now become widely adopted, to the extent of there now being a wide range of commercially available experimental apparatus available to perform such experiments compared to the situation 10 or more years ago when the vast majority of devices were home-made. SME techniques (figure 1) are being unleashed on exciting questions across science, particularly in understanding biological materials. A significant historical development in SME approaches from the last millennium came with the application of scanning probe methods, first utilized in scanning tunnelling microscopy (STM) to map surface topography [9] in 1982, followed by its use in non-biological condensed matter physics for manipulating single atoms [10] in 1990. In 1986 atomic force microscopy (AFM) was invented [11] and the trapping of dielectric particles with a light beam was achieved [12]. In 1991, far-field light microscopy was used to track gold nanoparticles attached to DNA being transcribed by immobilized RNA polymerase, and the following year the elasticity of DNA was probed with magnetic and hydrodynamic force [13]. In 1996 fluorescent imaging of single molecules in diffusion was achieved [14] and the structure of green fluorescent protein (GFP) was solved [15]. Single molecule detection was achieved via surface-enhanced Raman scattering (SERS) in 1997 [16], and in 1998 far-field microscopy was used to resolve numerous single molecules, breaking the Abbe diffraction limit, in three dimensions [17]. Comprehensive reviews of the early histories of single molecule approaches exist, such as those on single molecule spectroscopy [18] and DNA mechanics [19].
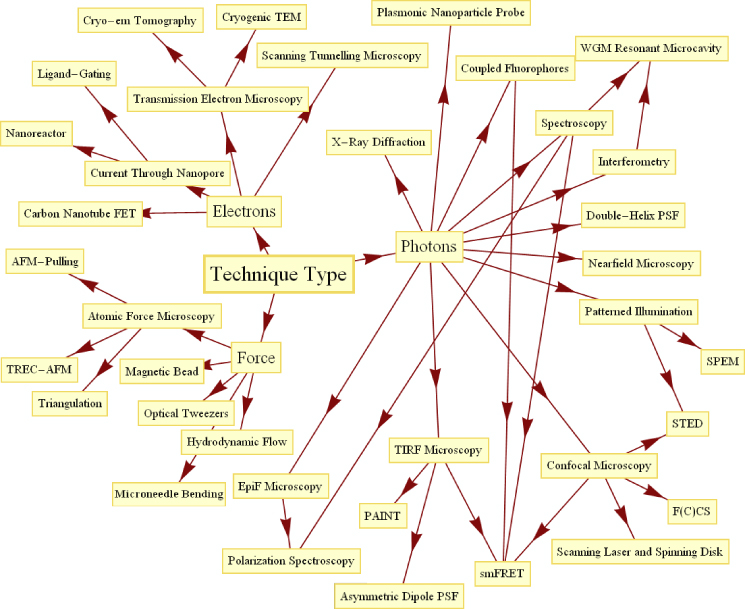
Figure 1. A technique-oriented network schematic of single molecule experimentation.
Download figure:
Standard imageIn the first decade of the new millennium, tremendous progress was made, including dramatic improvements in fluorophore, optical and data analysis technologies. What now exists is a range of techniques (succinctly represented in [1]) that have been aggressively applied in novel combinations to a diverse range of molecules and environments. The methods have been modified to become more biocompatible and gain ever deeper access to the molecular mechanisms in live samples. Biological insight has been gained with relevance to a number of topics including protein folding, membrane proteins, molecular motors and DNA–protein interactions, including the methods by which the DNA code is ultimately read off and converted into functional proteins via translation and transcription, respectively. The frontiers of scientific knowledge and experimental capability are now being challenged ferociously.
2.2. Single molecule systems
The purpose of the single molecule experiment imposes different expectations and requirements. In some cases the emphasis is on minimizing the perturbation to the molecular system of interest, while in other cases the fine control over environmental conditions is what allows the technique to ultimately be successful, and in many instances the ordering of the environment is the ultimate goal (for example, in nanotechnology). The focus of this review is on approaches that may conceivably enable one to gain specific information on biomolecules, in or out of their native contexts.
Where the effort is to characterize the molecules as independent entities, the default approach is to make measurements in the most reduced practicable environment. This may involve cryogenic temperatures, vacuums and spatial constraint; to minimize phenomena such as instrument noise, background artefacts, Brownian motion and contamination. At low temperatures, high-resolution optical methods can be applied and many more phenomena may be accessed [18].
In vitro methods allow for very specific physical characterizations of individual molecules with a broad range of probing techniques. Techniques can be used that might adversely affect the working order of a living cell. Small reconstituted systems of biomolecules may be composed in an attempt to reproduce molecular interactions and discover emergent behaviour that might be present in living cells [20]. The highly controlled and understood in vitro environment may make it more tractable to physically characterize the biomolecules and develop predictive, quantitative theories for their collective behaviour. This information can be considered in the interpretation of live-cell measurements, and vice versa.
In order to discover the true activity of biomolecules in their native physiological conditions, one must observe living cells. While environmental complexity may preclude full physical and chemical understanding, observations of phenomena may illuminate the biological mechanisms that make life possible. This has proved to be technically highly challenging for reasons that will be discussed later. Significant progress has, however, been made in the last 5 years, and some counterintuitive processes have been inferred. Imaging in time of a single molecule in a live cell is by its nature embracing the systems biology approach. The observed molecule is exposed to the numerous interdependent reaction networks as their activity changes to modify the organism's behaviour. Many molecules have been observed (including signalling molecules and transcription factors) whose activity is regulated by association with specific intracellular structures or by transport between subcompartments of the cell [21]. Analysis of the trajectories of individual molecules in the cell membrane has shown anomalous diffusion behaviour, a more hindered version of that associated with standard Brownian motion [22]. The line between what are the intrinsic and extrinsic features of a single molecule is blurred. Through its conformation, location and association within its environment, a single molecule can be a local reporter of the functional groups, atoms, ions, electrostatic charges and/or other sources of local fields in its immediate vicinity [18]. For example the structural form a protein holds in water is intimately related to hydrophobicity. A molecule's characteristics should be considered as a function of both its identity and its environment (figure 2). Viscosity has been determined by following the relaxation rate of DNA after end-to-end stretching and photoscission [23]. Solvent polarity and the pH of an aqueous medium can affect fluorescent properties. Using ratiometric measurements, ion concentrations can be robustly calculated [24].

Figure 2. The molecule under study exists as part of a larger experimental system, with both a physical and functional context.
Download figure:
Standard imageA theme of cell biology is that there can be paradigmatic similarity in the molecular mechanisms of cells across the diverse range of living species. This leads to the use of model organisms that can become well understood and are seen to give insight into a much broader range of species. An example that is a frequent subject of SME is the Escherichia coli bacterium (cultivated strains of the commonly used 'K12' variant of E. coli can be grown easily and are well adapted to the laboratory environment). Examination of such specimens and their constituents is currently being done according to two grand aspirations. The first is to understand cellular behaviour in a deterministic manner, based on biophysical analysis of constituent molecules. The second involves a more inclusive approach that sees complete understanding of the cell to be reliant on consideration of the entire dynamically interrelated system of molecules.
Soft condensed matter is defined as being characterized by weak interactions between polyatomic constituents, by important thermal fluctuation effects, by mechanical softness and by a rich range of behaviours [25], and this can be said of biomatter. Molecules exist within a milieu of fluctuating interactions including electrostatic forces associated with charges and dipoles, transient hydrogen bonds, dispersion, hydrophobic and osmotic forces. The well-known self-assembled tessellating structures of soft condensed matter include micelles and membranes, both of which are prevalent in biological systems and enable them to survive. An example of an area of active research is phase behaviour observed in the membrane [26–28].
2.3. What characterizes a living system?
With consideration of a system that is living, new themes and interactions become relevant. Living systems are distinctive in their use of free energy to create order, essentially functioning as machines that, on a local level, generate negative entropy changes. Non-equilibrium states are osmotically maintained through pumping of ions by molecular machines. Energy is obtained from light and food and molecular machines use it to build structures, to power actuators, to actively transport molecules, to generate heat and light and to regulate these processes. In their native environments, biomolecules have functional/logical significance as well as standard physical descriptions. A key aspect of biological systems is the specificity with which constituent molecules interact. Reaction rates are increased by enzymes with highly specific binding, transitions in macromolecules are sharpened by cooperativity, and biased random walks are attained based on the geometry of mechanisms [29]. Manifold heterogeneity is a fundamental characteristic of life.
The physical single molecule understanding we gained through SME may be many levels removed from the functional meaning that is of most relevance to the organism. Single molecules may exist in a highly complex molecular system in order to ultimately affect a basic mechanical property at the organism level, such as cellular stiffness [30]. Also, the nanometre length scale form of biomatter may only make sense in the light of the macroscale capabilities of the organism [31].
2.4. The central importance of fluorescence techniques
With the conjugation of a discernible label, a vast range of single molecule approaches become available. Labels have been developed synthetically and from naturally expressed molecules such as the now ubiquitous GFP. Various classes of label exist including plasmonic nanoparticle probes, fluorescent labels, quantum dots and chemiluminescent labels (in which the excited state is populated via a coupled chemical reaction) [32, 33]. Stimulated emission can also be employed in the imaging of non-fluorescent chromophores [34].
Fluorescent probes are designed to localize within a specific region and or associate with a specific molecule. Fluorescence is used as a mechanism for high sensitivity measurement because of the inherent 'Stokes shift'. Emitted light can be isolated from the excitation light as it has a maximum intensity at a longer wavelength. Fluorescence enables the detection of otherwise invisible molecules and the fluorophore may be re-excited many times to achieve a high signal within a given time. Fluorophores are characterized by a number of measurable properties including extinction coefficient (M−1 cm−1), quantum yield, and time to bleach (to half intensity at an excitation power that initially caused 1000 photons s−1) [35].
The phenomena of quenching (loss of fluorescence) can be a result of short range interactions with other fluorophores and the molecular environment including binding to a target molecule. In quenching, the fluorescence quantum yield is reduced without a change in the emission spectrum. Binding of a label to its target can reduce the fluorescent quantum yield and this can be used to monitor enzymatic cleavage [32]. Similarly, many fluorophores (that have a quenching effect on each other) may be attached to a molecule and an increase in brightness observed upon molecular cleavage. Another mechanism, Förster resonance energy transfer (FRET), is the non-radiative energy transfer between an acceptor–donor molecule pair that results from their dipole–dipole coupling when brought into close proximity (often the FRET pair of molecules are fluorescent and so some researchers refer to fluorescence resonance energy transfer synonymously). The visible result is that the emission peak of the fluorophore accepting energy increases in intensity.
Fluorescence based techniques are advantageous for live-cell measurement as they are relatively non-invasive, quantitative and can be recorded over 'real-time', i.e. a timescale which is at least as fast as the biological process under investigation. The ambitious goal of fluorescence microscopy on live cells is to be able to simultaneously discern multiple types of molecules in motion through time, in the three spatial dimensions, and in any region of the cell. Ideally one would be able to count and characterize the distribution of molecules in the cell and make inferences about the conformational state of each.
There exist significant challenges to the use of fluorescence for observations in living samples, indeed fluorescence imaging can have detrimental effects on the normal activity of a live cell. An unintended consequence of fluorophore illumination can be the production of reactive oxygen species (ROS) which are known for their phototoxic effects [36]. Some fluorescence imaging techniques including those that used photoactivatable fluorophores use relatively large illumination doses and high fluorophore concentrations and the effect on cell physiology should be carefully observed. The illumination dose necessary for imaging a stationary fluorophore has been shown to be reducible by spatially modulating the intensity [37]. While the intensity of light striking the edge of the observed fluorophore is retained, the light intensity striking both the centre of the observed fluorophore (where the signal-to-noise ratio, SNR, is high) and the dark background (where the SNR will never be high) is reduced. Even with standard non-photoactivatable fluorescence microscopy there is an issue of generation of phototoxic free-radicals, which ultimately limits the maximum duration of observation of a living sample. However, this effect is strongly wavelength dependent, getting worse at shorter wavelengths. To a certain extent this is being alleviated by the application of fluorescent probes that are excited towards the red end of the visible light spectrum or even into the near infrared.
In samples of living cells one must consider carefully the strategy for introduction of the labels. In order to bypass the cell envelope, a variety of strategies are available, including microinjection and protein transduction [38], though such methods work poorly for bacterial cells which are commonly used as model experimental organisms. A more universally applicable and elegant approach uses modified cell strains in which several key proteins can be expressed as fluorescent protein (FP)–fusion constructs. The genomic encoding approach has been shown to produce fluorescently tagged protein at levels which are comparable to the unmodified cell strains [39]. This contrasts with the more common approach of plasmid expression that would produce significantly more protein than would occur naturally. Due to the size of the FP tag being comparable to that of the protein under study, there is often impairment of functionality (50% being not atypical), depending upon the system and the specific measure of functionality. However, the advantages of using single molecule fluorescent tags (with in effect 100% tagging efficiency) generally outweighs the costs of such physiological impairment.
Careful selection of the FP is important [40]. The FP should express efficiently and without toxicity to the system under investigation [41]. It should be bright enough to be distinguishable from the background autofluorescence and should be sufficiently photostable to emit for the required excitation exposure. When two or more types of molecules are labelled there should be minimal cross-talk in the excitation and emission spectra of the FPs used (this is not the case in FRET investigations in which case we wish to maximize donor–acceptor FP spectral overlap).
Ultimately measurement is often made in a low SNR regime, which sets a limitation on the extent of processes which can be investigated simultaneously. The brightness of an FP recorded by the microscope's CCD camera is dependent on a number of variables. These include the intrinsic FP brightness, the optical properties of the imaging system (excitation wavelength and intensity, and the transmission spectra of components such as filters and dichroic mirrors used), and ultimately the sensitivity (dependent on wavelength) of the CCD itself. At a basic level this requires that the spatial and temporal resolution be high enough. This means that one must carefully consider the balance between sensitivity of detection, speed of acquisition and viability of the specimen [42].
Fluorescence microscopy in live cells has been used to observe gene expression and transcription, translation and replication dynamics in real-time [43]. Protein molecules have been shown to be produced in 'bursts' with copy numbers following a geometric distribution [44]. The regulation of gene expression was investigated by observation of transcription factor binding to DNA [45]. It was possible to infer the proportion of time a repressor protein (in the well-studied 'lac operon' model bacterial system of gene expression) spends diffusing along and non-specifically bound to DNA. Methods have been developed to fluorescently label RNA, previously elusive due to its high mobility and lack of long-term stability in the living cell. Results showed RNA to be non-uniformly distributed within the cell and to be localized in apparently ordered helical structures along the cell axis [46].
3. The detection of single molecules
The structure of this article is based on the premise that the primary interest is in the underlying set of scientific questions, and the methods we devise to address these are necessary by-products to addressing such questions, as opposed to being an end in themselves. In this case, a fresh approach may be to consider the type of information one wants, and then appraise a broad range of experimental techniques that could conceivably be used (even if in reality certain details preclude use of a subset of the conceivable methods). So a series of general questions are asked relating to the capabilities of SME, and an attempt is made to answer each with a varied range of specific examples of successes from the literature (figure 3). The questions are roughly ordered in increasing levels of sophistication as to the type of information obtained or manipulation achieved.

Figure 3. A capability-oriented network schematic of single molecule experimentation.
Download figure:
Standard image3.1. Is a single molecule present (or absent)?
The presence/absence of a single type of molecule may have a pronounced effect on the system of which it is a part. For example, an inhibiting effect on a chemical pathway, an abnormality in a gene knock-out mouse, or more simply a characteristic colour. But in these cases, there is no knowledge or control of the number of those molecules, no direct quantitative measure on a single molecule. Observation of such effects would not be classed as single molecule observations.
Biomolecular mechanisms may indicate the presence of a single molecule. The presence of a single molecule (e.g. ATP) could conceivably be evidenced by the energy it transfers to a molecular motor, though with one single processive step may be impossible attribute above the thermal noise. The presence of a molecule might be inferred from the effects on the conformation of a second molecule such as a membrane pore, pump or receptor. If the single molecule gates an ion-channel, it may have an effect (that is generally stochastic [47]) on membrane resistance. The validity of using this mechanism as a 'single molecule assay' would depend on whether it is a specific type of single molecule that activates the membrane pore. Presence could be inferred if the molecule were to open/close the pore for a long enough time for an observable and significant conductance change. The first measurements of single ion-channel conductance changes were achieved via patch clamping in 1976 [48]. Ion channels may be gated by voltage, pH and the presence of ligand molecules [49] and recently have been integrated in modern bio-electronic devices [50].
A charged single molecule might be registered through direct contact with a detector electrode. In this case the detector would require high sensitivity and an absence of noise, requiring highly controlled environmental conditions, and the likely cooling of the electronics in order to reduce dark current.
If all the molecules of a sample are spatially constrained in a sparse monolayer on a surface, an image can be constructed. A technique with molecular resolution will make evident the presence of a molecule and might enable some form of identification. Popular microscopic techniques capable of resolving molecules include transmission (TEM) and scanning electron microscopy (SEM) and the scanning probe methods of near-field optical microscopy, scanning tunnelling microscopy and AFM. The applicability of these methods depends further on the properties of the molecules and the environment.
Simultaneous topography and recognition imaging (TREC) uses an AFM with a cantilever tip carrying a ligand molecule [51]. Through specific binding to molecules of interest, and the associated decrease in cantilever oscillation amplitude, simultaneous measurement of topography and molecular recognition is achieved [52].
More technical capabilities become available with molecule-specific spatial constraints. For this, an attachment/confinement mechanism is required, though it does not necessitate labelling the molecule with an observable molecule or nanoparticle. If a molecule can be probed without the conjugation of a label, several potential problems may be avoided. An adequately specific label may be hard to find, and conjugation may be difficult. Once attached, it may sterically hinder the molecule of interest and interfere with its conformation.
Through some form of attractive or confining mechanism, a single molecule in solution may be immobilized at a known position such as on a latent pattern on a substrate surface, or the surface of a probe. Recent work used sequential adsorption of protein A and IgG to a silver nanoparticle micropatterned surface [53]. Such high ordering of a pattern can enable the separation of a SERS-enhanced spectrum (note the review [54]) of the biomolecules of interest, from that of the substrate and non-specifically adsorbed species. This approach allowed detection of biomolecules down to almost 500 fM concentrations.
Silicon nanowires arrays have been used to detect DNA molecules at a concentration limit of 10 fM [55]. Here, the nanowires were functionalized with peptide and nucleic acid, and resistance changes were measured upon hybridization with the DNA. More recently, single molecule detection of DNA hybridization was achieved with a carbon nanotube field effect transistor [56]. Conductance changes were measured when a diffusing strand of DNA hybridized with a complementary strand that was covalently bound to a point defect in a carbon nanotube.
Two ensemble optical techniques are used to register the addition of molecules to a surface through interaction with an evanescent field. Dual polarization interferometry (DPI) works by virtue of the change in polarization of the field, and the surface plasmon resonance sensor (SPR) works by virtue of the resonance shift of electrons at the interface between the surface and a thin metal layer. In 2007, SPR was advanced to reach single molecule sensitivity through the incorporation of a 'whispering gallery' microcavity [57]. This form of sensor has been shown to have potential for a multitude of further capabilities [58].
In the simplest case, a detected molecule may be identified through knowledge that it is of the only species present. A number of ensemble techniques spatially separate the molecular species in a sample then make measurements of respective abundance. Separation has been achieved through a range of mechanisms by virtue of molecular mass, size and shape. Molecules may be separated into sedimentation bands within a density gradient in an analytical centrifuge. In dialysis, the solvated sample is filtered through membranes of set pore sizes. In chromatography methods, the sample is introduced to a medium in which there is a stationary and a transient phase and molecules are separated based on their flow rate. In conventional mass spectrometry, an ionization process creates fragments of molecules with different mass-to-charge ratios. The fragments are then separated through the differential effect on their trajectories as they pass through a magnetic field.
Alternative forms of mass spectrometry have been approaching single molecule sensitivity. The vibrational frequency of a nanoelectromechanical resonator is exquisitely sensitive to small variations in mass. This has been made use of in a novel form of mass spectrometer, to infer the adsorption of molecules in real-time [59]. The approach is theoretically capable of mass sensitivity below 1 Da. Another paradigm for mass spectrometry was to use a single membrane pore and observe the effect on electrical conductance [60]. The residence time of a molecule in a pore may increase monotonically with its mass and have a measurable effect on ionic current.
3.2. How many single molecules are there?
The ability to robustly quantify how many biological molecules are involved in any given process, for example how many sub-units really constitute a functional complex in a molecular machine, is vital to our understanding of the real molecular mechanisms involved in living soft condensed matter. Single molecule methods excel in determining molecular-level concentrations, particularly when the sample is too small for an ensemble method, as in the case of a cell where the copy numbers of many proteins are very low. In many cases, the number of pathogenic molecules necessary to establish disease is very low, and there has been much work to develop techniques capable of measuring smaller and smaller concentrations as a result.
Counting can be seen as an extension of registering the presence of a molecule type. At some level, there must be a reasonable confidence that it is counting and not kinetics, for example, being able to distinguish between repeat signals from the same molecule over time. If measurements are made simultaneously there must be some way to distinguish the separate signals from the different molecules. Measurements made over extended periods of time require some mechanism that rules out a molecule from repeat measurement (e.g. photobleaching).
For simultaneous counting, imaging is an obvious choice, though molecular motion during the capture period must be ruled out. This could be done by fixing all molecules to a surface, though this may take the system far from its native physiological state. If the image is analysed as a single channel recording (as AFM or SEM micrographs, when interpreted as purely topographical depictions), the resolution must be high enough for the number density of molecules. If the molecules are large enough to be visualized no labelling is required. For type-specific counting, molecules may be identified by the knowledge that they are the only species present, or based on their form.
In the established ensemble method of spectrophotometry, the transmission of light through a sample is measured as a function of wavelength from which the concentration can be inferred. Mean reactions from populations of several thousands of molecules can be observed also, where dyes are present whose fluorescent activity changes upon binding to specific chemicals.
With the use of a fluorescent label, measurements of sub fM concentrations have been achieved [61]. Low abundance proteins were captured with antibodies on microscopic beads (with low likelihood of more than one protein per bead according to Poisson statistics) and labelled with an enzymatic reporter capable of producing a fluorescent product. The beads were then isolated in arrays of femtolitre-sized wells, into which no more than one could fit. Because of the resulting confinement, a high fluorescent signal was observable from those wells in which the proteins of interest were present. The percentage of wells showing fluorescence was then taken to give a measure of concentration. This approach enabled protein concentrations of about 10−19 M to be measured.
In fluorescence correlation spectroscopy (FCS), analysis is made of fluctuations in the fluorescence intensity from diffusing labelled molecules within a confocal volume [62]. From this it is possible to determine the localized concentration and molecular mobility. Any change that affects a molecule's mobility may be picked up by FCS. With fluorescence cross-correlation spectroscopy (FCCS), one can determine common features in signals from two distinct fluorescence channels. This may enable the proportion of particles that are double-labelled (i.e. comprise two bound molecules) to be determined. The fluctuations observed in FCS are more pronounced, the lower the mean number of molecules. This means that the upper bound of concentration is limited by the size of the confocal volume, and accordingly attempts have been made to produce smaller observation volumes. Two-dimensional arrays of zero-mode waveguides, essentially small holes in a metal film deposited on a microscope coverslip, have been used to this effect. Beyond a cut-off wavelength no propagating wavelength modes may exist and illumination is evanescent from the base of the waveguide. This has the effect of delimiting to sub-wavelength all three spatial dimensions of the excitation volumes [63].
Counting of molecules in cells brings new challenges. Stochasticity exists in the expression of constituents resulting from intrinsic and extrinsic noise [64]. One approach is to lyse the cell and analyse the constituents in vitro, possibly using a method described above. This will fail to pick up the heterogeneities in the spatial distribution of molecules in an active cell, but may at least allow for a full accounting of raw molecule numbers to be carried out. Micro- and nano-fluidic 'lab-on-a-chip' devices [65] can potentially combine the processes of manipulation, lysing, fluorescent-antibody labelling, separation and protein quantification for a single cell. The single molecule pull-down approach may enable isolation of complete protein complexes in vitro for examination through fluorescent methods [66].
Ideally a non-perturbative approach with high time resolution would count molecules in regions of living cells. The phenomenon of photobleaching has been taken advantage of in order to count the number of molecules within an active membrane-bound complex in a living cell [39]. Using TIRF microscopy a video was taken of GFP-labelled proteins (in vivo) involved in force generation in the bacterial flagellar motor. Analyses were made of a region of interest encompassing immobilized flagellar motors. Intensity was measured with time and exponential photobleach decays were observed with step-like behaviour visible near the tail of the measured intensity versus time traces. The underlying periodicity in the intensity was examined in a power spectrum of the pair-wise difference distribution of each bleaching curve. A fundamental peak was then identified as a possible indicator of the intensity of a single fluorescent protein. Dividing the initial total intensity of the region of interest by this value gave an estimate of the number of fluorescent proteins present in the motor.
Recently a yellow fluorescent protein fusion library of E. coli was generated with 1018 different proteins labelled for the first system-wide analyses of expression [67]. A microfluidic chip was used with fluorescence imaging of 96 strains in parallel, to quantify the protein and respective mRNA presence with single molecule sensitivity.
4. Investigating intramolecular characteristics
4.1. What is the structure of a single molecule?
Biomolecules typically have many levels of organization that result from environmental conditions and the sequence of constituent atomic groups. The form of a protein is generally that which minimizes free energy and results from the order of its amino acids. Numerous ensemble techniques have been applied to determine molecular structures. Circular dichroism, the differential absorption of left and right circularly polarized light, has been used to indicate the presence of chirality and relative proportions of different molecular structures. Such investigation has achieved single molecule sensitivity [68], revealing strong intrinsic circular dichroism responses and indications of molecular orientation. In the presence of an external magnetic field, magnetic nuclei will resonate between spin states upon absorbing electromagnetic radiation of a frequency that is dependent on their local chemical environment. Through nuclear magnetic resonance (NMR) spectroscopy, it is possible to obtain separate signals from each atom in a protein that can be characterized by frequencies that provide both distance and orientation constraints for structure determination [69].
Diffraction investigations such as x-ray, neutron and electron diffraction of crystalline samples have revealed the atomic structures of many biomolecules. However, there exist many species that have not been successfully crystallized. An ordered ensemble of molecules present as a crystal has been necessary to gain enough signal before the sample is destroyed by the impinging beam. However, theoretically it was believed that if a pulse of x-rays was intense enough, it could generate an adequate signal from a single molecule before the molecule exploded due to heating. It was recently shown that it should be possible when a purpose built x-ray laser (the most powerful in the world) was shone into a mimivirus to achieve a reconstruction with a spatial resolution of about 32 nm [70]. With higher irradiance pulses and detectors with higher dynamic range, it is foreseen that the structures of single molecules may be produced by such an approach.
With electron microscopy, structural features in fixed biological samples may be visualized. A contrast agent is added to the sample that associates preferentially in different parts of the sample and structure is revealed as electrons are variably transmitted (TEM) or scattered (SEM). In cryogenic electron microscopy (cryo-EM), the rapid freezing to cryogenic temperatures can allow biological samples to survive the vacuum conditions necessary for TEM without the need for fixing. Standard cryo-EM with single particle reconstruction (where many similar particles are imaged and correlated) has been able to produce 3D data of a high enough resolution to be fit with an atomic model [71]. With cryo-EM tomography only one particle is examined, and 3D structure reconstruction is produced from images at a series of orientations around its centre [72]. A strength of cryo-EM is the capability to generate maps of the relative locations of many proteins in a complex. Higher resolution molecular structures generated from crystallographic methods may be used to interpret the tomographs with less ambiguity.
The basis for a single molecule approach to determining protein structure has been developed that makes use of mechanical triangulation [73]. With prior knowledge of the protein sequence, angstrom-precise respective positions of three amino acids in GFP were found in solution. The process involved measuring the change in distance between each pair of the three acids before and after pulling them apart to a completely stretched conformation. Several approaches are now being explored, taking advantage of the constricted geometry of pores to infer the bases of a DNA molecule [74]. Other single molecule sequencing approaches are being developed including 'sequencing by synthesis' and with TEM [75].
4.2. Can we determine the conformational state of a single molecule?
As well as being affected by the incessant Brownian barrage, the conformation of biomolecules develops with folding from the initial state of synthesis, and may change further upon binding (in allosteric molecules). The functionality of biomolecules is largely based on their 3D form and the energy landscape that guides conformational change [76].
FRET is employed for high precision measurements of inter-fluorophore distances of up to about 10 nm. FRET is made use of in two main configurations, the most relevant to conformational measurements being where one molecule is labelled with the donor and acceptor [77]. The second configuration is where one of two molecules that may associate is labelled with a donor, and the other an acceptor. This is used to observe binding. Configurations that involve more than two fluorophores per molecule are also being developed [78].
Using alternating-laser excitation (ALEX) of single molecules, it is possible to characterize both intramolecular distances (based on donor, acceptor FRET) and to infer whether a molecule is labelled with a donor, acceptor or both to give a measure of stoichiometry. This can be used in the spectroscopy of molecules diffusing through a confocal volume, or in microscopy of immobilized molecules on a surface [79].
Denaturation, the disruption of the non-covalent intramolecular bonds, of molecules in an ensemble, can be achieved with solvents or high temperatures. At a single molecule level conformational changes may be induced and observed through methods of force spectroscopy. Techniques that probe with force include optical tweezing, magnetic tweezing, AFM, glass microneedle manipulation and flow-induced stretching. Force-based methods enable manipulation over six orders of magnitude in length (10−10–10−4 m) and force (10−14–10−8 N) [3], though typically the scales relevant to biomolecules are the nanometre and piconewton. Direct mechanical measurement of elastic properties of single molecules can be made, for example on DNA [13], and to observe the metastable and transient folding states on large molecules such as GFP [80]. Through combining optical tweezing and FRET, improvements in force sensitivity have been achieved [81].
A complementary approach to force-based SME is that of steered molecular dynamics. This is where in silico molecules are probed as in vitro experiments, with the aim of gaining deeper insight into the mechanical mechanisms at play [82]. Coarser modelling approaches have been developed for understanding features such as anisotropic deformation response of molecules over times scales closer to those of force-based experiments [83].
5. Investigating intermolecular details
5.1. The orientation of a single molecule
Linear dichroism is an ensemble polarized spectroscopy technique that can provide information about relative orientations of sub-units of biomaterials and orientations of the whole biomaterial with respect to an orientation axis [84]. The orientation of molecules in a cuvette affects the degree of absorbance of light linearly polarized parallel and perpendicular to an orientation axis. Polarization spectroscopy of single fluorophores has been used to probe rotation of surface immobilized molecules and the polarization anisotropy of freely diffusing molecules [85]. Recently, single molecule fluorescence polarization made evident a continuous helical rotation involved in biofilament elongation and depolymerization [86]. Fluorophore dipole orientation can also be determined from a real space image through maximum-likelihood fitting of an anisotropic diffraction limited spot [87].
It is often useful to control the orientation of a molecule. In ensemble linear dichroism, elongated molecules may be co-oriented by shear forces produced by rotation of a cylinder within the measurement cuvette. Standard methods of single molecule orientation include tethering a molecule to a surface at one end and exerting a hydrodynamic force, or magnetic force, on a magnetic particle attached to the other end [13]. Torque may be applied directly to the magnetic particle, as is the case in experiments on the super-coiling of DNA [88].
5.2. Single molecule spatial location
The location of a single molecule could conceivably be determined in several ways. Triangulation could be based on a quantity (such as magnitude of irradiance) that monotonically changes with distance from the molecule. The molecule could be located spectroscopically by the effect of the environment, if for example fluorescence emission changes in a way that has been pre-mapped. Likewise, the response may indicate location with illumination or probing that varies with position in some manner.
The standard approach is to create a map of the area or volume, and in it the molecule is evident. This can be done with a scanning method or by a far-field method. If the molecule is constrained to a known geometry such as a flat surface or a bacterial membrane, less information may be necessary to infer its location. Reducing the regions of probing or illumination is a design principle across a number of SME techniques including laser scanning confocal microscopy (LSM or LSCM), scanning near-field optical microscopy (SNOM/NSOM) and total internal reflection fluorescence (TIRF) microscopy. In SNOM/NSOM light is sent through a sub-wavelength optical aperture in a transparent metal coated probe tip that scans sample topography. This produces illumination of a surface region that is laterally smaller than that in confocal excitation [89].
There is a commonly employed method that gives typically an improvement of about ten-fold in the precision with which one can locate an isolated object whose characteristic length dimensions are less than the diffraction limited optical resolution of a far-field imaging system. The method, applied in localization and tracking contexts, involves calculation of the centroid of the two-dimensional Gaussian function of the spatial distribution of fluorescence intensity from the object, which is fitted over the object in the image. The precision of the calculated centroid is dependent on the number of photons emanating from the fluorophore [22]. Noise affecting the precision comes from the fluorophore (in the form of 'shot noise'), and the background (a result of factors including out of focus fluorescence and readout noise of the CCD), as well as from uncertainty of where precisely a detected photon was incident within a given CCD camera pixel (so-called pixellation noise).
TIRF microscopy is an approach that allows for the excitation of a broad but very shallow region of space beyond a cover slip. A beam emerging from the edge of the objective lens, passing through oil and entering the cover slip, strikes the sample surface of the slip at an angle that results in total internal reflection. At this point an 'evanescent' field is produced with a 1/e penetration depth of typically ∼100 nm (normal to the surface and into the sample) [90]. This field is useful as it can be used to excite fluorophores within a cell's membrane with minimal excitation in other regions, dramatically reducing the noise due to scattering and fluorescence of molecules in other parts of the cell and of the surrounding buffer. TIRF microscopy has made it possible to discern the presence of, and track, single membrane proteins above the usually prohibitive noise [91].
TIRF microscopy and the centroid calculation localization process were combined and called fluorescence imaging with one nanometre accuracy (FIONA) [92]. The method was famously used to examine the step lengths of myosin molecules (labelled with single Cy3 dye molecules) moving along actin filaments in vitro. Analysis of the processive step lengths was seen to indicate the relevance of a particular 'hand-over-hand' model for myosin motion.
Alternative approaches use additional patterned illumination of the sample to affect fluorescent yield and obtain higher resolution. One technique inhibits fluorescence in the outer regions of the illumination point spread function through stimulated emission (STED) [93]. In saturated pattern excitation microscopy (SPEM) higher resolution is obtained from the generation of higher spatial harmonics in the emission pattern. These result from the nonlinear dependence of fluorescence on illumination intensity when fluorophores are saturated with standing wave illumination [94].
The point spread function of a standard microscope gives little information on the axial position of a fluorophore and in a diffraction limited system the resolution is typically three times poorer than in its focal plane, though various ingenious optical techniques have been applied to reduce this [95]. Techniques including astigmatic and biplane imaging and use of a double-helix point spread function have achieved axial location precision of under 10 nm [96].
5.3. The relative position of many single molecules
The location of many fluorophores within a diffraction limited spot ('super-resolution imaging') may be attempted simultaneously or sequentially. When done simultaneously, an extra dimension of discrete channels must be used to isolate the signal from each fluorophore. The standard way to achieve this is by using fluorophores of a variety of colours. A two-colour version of FIONA, single molecule high-resolution colocalization (SHREC), was developed capable of inter-fluorophore measurements below 10 nm in resolution [97]. The molecule of interest is double-labelled with two chromatically different fluorophores and excited with lasers corresponding to the excitation spectrum of each. The emission is separated with dichroic mirrors and filters and directed to differing positions on a CCD. A mapping function is then used to correlate the images and determine the inter-fluorophore distance.
Fluorophores may be imaged sequentially through the ability to have different fluorophores fluorescing at different times. This may involve photoactivation, photobleaching or reversible photo-switching, termed single molecule active-control microscopy (SMACM). Sequential imaging may also be achieved through approaches such as STED that induce fluorescence from sub-diffraction limited regions [98]. In points accumulation for imaging in nanoscale topography (PAINT) super-resolution images of surface structure are generated from transient binding and associated dequenching of fluorophores diffusing from solution [99].
Photobleaching has been made use of in techniques to improve spatial resolution and reduce background fluorescence, aiding the tracking of molecules. Single molecule high-resolution imaging with photobleaching (SHRImP) is a technique that takes advantage of the quantal nature of photobleaching to resolve two fluorophores separated by distances down to 5 nm [100]. A movie is made of two fluorophores moving in close proximity to each other. The total intensity is recorded until one molecule photobleaches and a stepwise intensity decrease is observed. The remaining fluorescence is said to come from the one remaining fluorophore and a two-dimensional Gaussian distribution is fitted to its image. This allows the (FIONA) localization of the remaining spot to a precision of ∼1.5 nm. The previous frame is then observed in which both fluorophores are photoactive and the single fluorophore Gaussian distribution is subtracted from the total to determine the intensity distribution of the remaining fluorophore. Comparison of their localization allows for the high precision distance measurement. It should be remembered that the distance measured is only the image plane two-dimensional projection of distance.
In some fluorophores a saturable transition can be induced between two conformational states, by using much lower intensities of light than are necessary for rapid bleaching. This mechanism has effectively been used to switch fluorophores on and off as a basis for super-resolution imaging [101]. There has been much interest in techniques that use this process to locate densely distributed biomolecules. In photoactivated localization microscopy (PALM) [102] photoactivatable fluorescent proteins are used, all of which are initially in their inactivated state. Then the sample is irradiated for long enough to activate a sparse subset which is imaged until bleaching occurs. This process is repeated until all fluorophores have been activated, imaged and bleached. Summing the frames yields an image with resolution of 2–25 nm.
In sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) [103], fluorophores are switched between a fluorescent and dead state in a reversible manner by light of different wavelengths. The process involves going through cycles where only a fraction are activated and imaged in order that (as with PALM) the population appears sparser than it is. After a sufficient number of cycles, the images are correlated to map the position of all fluorophores. Originally, organic fluorescent dyes were applied in vitro for STORM investigations [103]. Next, the process was extended to multiple colours [104]. In 2008, the method was extended into the third spatial dimension giving image plane resolution of 20–30 nm and a depth resolution of 60–70 nm [105]. High quality super-resolution imaging of molecules in a living cell is now possible with fluorescent proteins [106].
Two new labelling approaches may have significant impact on in vivo imaging. Developed for use in PALM type imaging investigations, HaloTag-based target-specific labelling enables the use of bright organic fluorophores as an alternative to fluorescent proteins [107]. A fluorescent flavoprotein has also been developed that ultimately enables correlated electron microscopy [108]. Live fluorescent images of the genetically encoded protein may be taken, and then higher resolution TEM images of the fixed cell may reveal finer structural details.
5.4. Single molecule translational activity
An extension of localization and imaging is the tracking of molecules over time. This requires that the molecules be repeatedly localized within a time window that is short enough to resolve the dynamics of interest. It is attractive to carry out motility experiments in vitro because one need not consider the effects of high fluorescence backgrounds, complicated filament geometries, non-uniform drag coefficients and other enzyme activity [109]. One may also modify the environment to regulate molecular activity. In order to resolve in time the hand-over-hand steps of myosin V, the concentration of ATP (the molecular motor fuel) was reduced to slow dynamics [92].
Force-based methods have also been used to monitor the motion of processive molecular motors. The transcription process in which RNA polymerase moves along DNA was monitored through conjugated beads held in optical tweezers. The length of discrete processive steps and force–velocity relations were obtained [110].
In living cells, analysis is often aimed at characterizing the motion of a molecule to infer whether it was freely diffusing, or had some sort of biased motion indicative of being involved in a biological process such as active transport. Ensemble molecular diffusion can be characterized by mean squared displacement against time interval, where free Brownian motion is inferred from a linear trend and anomalous diffusion otherwise. Single molecule tracking data may give access not only to the mean, but to the full probability distribution of square displacements [111]. Statistical tests have now been developed to distinguish between random, confined and directed modes of motion in single tracks, and have been applied to the interpretation of three-dimensional in vivo mRNA tracks obtained using double-helix PSF microscopy [112]. Where the mode of diffusion is already known, FCS may be used to extract the diffusion coefficient and other associated parameters.
In live-cell studies, techniques such as fluorescence recovery/loss in photobleaching (FRAP/FLIP) are used that make use of photobleaching to reduce background fluorescence. In FRAP, a region of interest in the surface of a cell is photobleached typically with a high intensity focused laser pulse. Afterwards, migration of photoactive molecules into the region can become evident and one can make inferences of the mobility of molecules in the membrane. FLIP is in many ways the inverse of FRAP—one makes observations of dark molecules entering a region from a nearby photobleached region. One study using these two methods revealed a counterintuitive dynamic turnover of stator units in the E. coli. flagellar motor [39]. Using modifications of the same techniques, proteins involved in respiration were investigated and appeared to be localized in mobile membrane-bound patches [113]. When the intermolecular distance is less than the optical diffraction limit, super-resolution methods such as those that use photoactivation or switching are used; however, such methods cannot currently be applied to simultaneous tracking of all labelled molecules due to the timescale of approximately seconds necessary for image generation being too long to resolve the dynamics that are typically of interest. However, such techniques may still be advantageous with a densely distributed population. Multiple sparse resolvable subsets of molecules may be tracked without photobleaching the remainder. The process can be repeated and many more tracks can be obtained than would have been possible otherwise [102].
5.5. Observing molecular binding and unbinding
Bonding characteristics can be explored through both mechanical probing, and monitoring of the binding kinetics. The bond strength of two molecules is a function of the free energy upon binding minus the sum of the free energies when unbound. This is directly related to the equilibrium constant of the binding reaction, which itself can be determined independently from the association and dissociation rates within a mixture.
The application of force to a single molecule can alter the free energy landscape for a reaction, affecting the thermodynamics and kinetics respectively [114]. The rupture force for an individual covalent bond was first directly measured through tension on a polysaccharide molecule bound end-to-end between a surface and an AFM probe [115]. Force–extension curves were produced in which rupture events were evident as kinks in the curve, giving rise to characteristic 'sawtooth' patterns when several rupture events are involved. Prior to this, the rupture force between two actin filament monomers was measured through the flexing observed in a glass microneedle bound in perpendicular orientation to one end of the filament [116]. The tension was applied from a relatively stiff glass microneedle attached in parallel orientation with the other tip of the filament. In the same study the technique was used to measure the ATP-dependent force exerted by myosin heads upon actin filaments.
With suitable time resolution, the techniques capable of molecular detection enable studies of kinetics. In an aqueous solution, binding kinetics can be monitored by the difference in appearance of a bound and freely diffusing fluorophore. In such a way ATP turnover was monitored with TIRF microscopy focused on the binding site of myosin [117]. FRET may be employed to study binding via two schemes. If the donor and acceptor fluorophore are on separate molecules, acceptor intensity will increase upon binding. The event may alternatively be monitored with both donor and acceptor on one molecule if there is an associated change in its conformation. Microfluidic devices make possible the miniaturization of flow-based assays and rapid combinatorial processing. Mixing and associated FRET measurements can be orchestrated at high rates, enabling measurement of very short lived states [118]. Control of the molecular nano-environment and the effect on binding is being explored through the use of membrane protein 'nanoreactors' [119].
The position of a molecule may also track kinetic activity, as in DNA synthesis where an increase in length results from addition of a base. Through two-colour TIRF microscopy, the rate of synthesis of flow-stretched DNA was recently monitored while simultaneously detecting polymerase association on the DNA [120]. Many further investigations have been carried out into protein–DNA interactions [121]. Within limited sample areas AFM can produce a video of topography with high enough spatial and temporal resolution to capture conformational dynamics. This was recently demonstrated in a study of the behaviour of torque-generating β sub-units in the stator rings of the rotary molecular motor F1-ATPase at a range of physiologically relevant ATP concentrations [122].
6. Perspectives
The single molecule methods applied to the living component of soft condensed matter have now entered the next generation phase. The first generation was characterized predominantly by in vitro experiments using one single molecule technique, generally on isolated, purified molecules divorced from their native context of the living cell. The next generation is characterized by far greater experimental complexity; as we have seen, it is now common to apply multiple single molecule techniques simultaneously in order to extract multi-dimensional levels of information from any given experiment. In addition, many in vitro experiments now include multiple molecular components to create a controlled biochemical environment much closer to the native context, with such developments going hand-in-hand with improvements in microfluidics design and implementation. Finally, it is now feasible, especially using advanced fluorescence microscopy techniques, to monitor soft condensed matter at a single molecule level in actual living cells in real-time, with improvements in super-resolution approaches permitting knowledge of spatial precision at, in principle, sub-nanometre levels.
The most likely improvements in the near future will be two-fold. One will involve improvements in sampling speed: this will necessitate the development of brighter, smaller, more stable fluorescent dye tags, more efficient dye delivery mechanisms into the living cell and improvements in camera speed and sensitivity technology. This may permit not only multi-dimensional levels of information to be extracted from any given single molecule experiment, but to do so in living cells over much faster sub-millisecond timescales than are available at present, which may ultimately give insight into the fast molecular conformational changes—the transient, unstable switches in molecular structural states—that give rise to many fundamental mechanical changes in the molecular machines that drive some of the most important biological processes in the living cell. The second level of improvement is likely to involve force spectroscopy on living cells. Currently, single molecule mechanical investigations are limited either to in vitro experiments or in vivo involving molecules which are accessible on the surface of cells (for example, protein complexes integrated into the cell membrane). However, all biological process at some level involve a force dependence between a molecule and a substrate, which is implicit if the process involves an interaction between the two. It is probable therefore that more research efforts will in future be devoted to optimizing single molecule techniques which might allow manipulation and measurement of molecular forces inside living cells as opposed to just on their surface.
Acknowledgments
This research was supported by a Royal Society University Research Fellowship (MCL), the EPSRC (OLJH and MCL) grant number EP/G061009, and a Hertford College Oxford Research Fellowship (MCL).