
Abstract
The interplay of spin–orbit coupling and Coulomb correlations has become a hot topic in condensed matter theory and is especially important in 4d and 5d transition metal oxides, like iridates or rhodates. Here, we review recent advances in dynamical mean-field theory (DMFT)-based electronic structure calculations for treating such compounds, introducing all necessary implementation details. We also discuss the evaluation of Hubbard interactions in spin–orbit materials. As an example, we perform DMFT calculations on insulating strontium iridate (Sr2IrO4) and its 4d metallic counterpart, strontium rhodate (Sr2RhO4). While a Mott-insulating state is obtained for Sr2IrO4 in its paramagnetic phase, the spectral properties and Fermi surfaces obtained for Sr2RhO4 show excellent agreement with available experimental data. Finally, we discuss the electronic structure of these two compounds by introducing the notion of effective spin–orbital degeneracy as the key quantity that determines the correlation strength. We stress that effective spin–orbital degeneracy introduces an additional axis into the conventional picture of a phase diagram based on filling and on the ratio of interactions to bandwidth, analogous to the degeneracy-controlled Mott transition in d1 perovskites.

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Electronic Coulomb correlations are at the heart of a variety of exotic properties in compounds with partially filled 3d or 4f shells. Prominent examples are found among the 3d transition metal oxides, where unconventional transport behaviors, ordering phenomena or unusual spectroscopic properties are observed [1]. It was argued early on that the comparably weak spatial extension of 3d orbitals leads to large electronic Coulomb interactions, competing with kinetic contributions. Depending on crystal fields, hybridisation, Hund's exchange, and band filling, this interplay can lead to renormalized metallic behavior such as in simple oxides like SrVO3 [2, 3] or iron pnictide compounds [4–9] or induce Mott insulating behavior like in YTiO3 [10] or V2O3 [11–14]. According to common belief held until recently, such effects would be less dramatic in 4d, and even less so in 5d compounds, due to the substantially more extended radial wave functions of those shells, as shown in figure 1. The discovery of Mott insulating behavior in Sr2IrO4 therefore triggered a little revolution in the field [15, 16]. In 5d oxides, spin–orbit coupling acts on an energy scale comparable to the other scales of the system (Coulomb interactions, bandwidths, ligand fields...), and the electronic state is the result of a complex interplay of Coulomb correlations, spin–orbit splitting and crystal field effects (for recent reviews, see [17, 18]). But, as pointed out earlier [19, 20], spin–orbit interactions can also influence the electronic properties substantially in 4d compounds. In Sr2RhO4, for example, the experimentally observed Fermi surface can only be reconciled with experiments when spin–orbit coupling and electronic Coulomb correlations are taken into account [19–22]. Here, we give a review of recent efforts to describe correlated spin–orbit physics from first principles, in a combined density functional and dynamical mean-field theory framework [21].

Figure 1. Radial distribution function as a function of the distance from the nucleus r expressed in atomic units, for the 3d, 4d and 5d orbitals. To ease the comparison between different atoms, we use the renormalized distance on the abscissa, where Z is the effective nuclear charge for a given multi-electron atom. As the principal quantum number n increases, Z remains almost constant for d valence electrons and their radial distribution is thus more and more extended.


Download figure:
Standard image High-resolution image2. Spin–orbit materials—an incomplete literature review
The term spin–orbit material refers to systems where spin–orbit coupling (SOC) and its interplay with other elements of the electronic structure—crystal or ligand fields, Coulomb correlations, magnetism,...—is essential in determining the physical properties. In many such materials, the physics is largely determined by the geometrical aspects of the crystalline structure, and the electronic properties can be understood by analysing the one-particle band structure. In particular, strong enough spin–orbit coupling can cause band inversions, possibly leading to non-trivial topological effects. The quest for topological materials is nowadays a hot topic of condensed matter physics, and several excellent reviews exist in the literature [23–25].
The scope of the present review is, however, a different one. Here, we focus on materials, where the interplay of spin–orbit interactions and Coulomb correlations is crucial, and the band picture is at best useful as a starting point for further many-body calculations. Early examples are found among the layered tantalum chalcogenides: TaS2 [26–28] is Mott insulating thanks to the presence of a lone narrow band resulting from the combined effect of SOC and a charge-density wave instability. The corresponding selenide, TaSe2, [29] displays a surface Mott metal–insulator transition. Nevertheless, the true power of the interplay of spin–orbit interactions was fully appreciated only after the discovery of Sr2IrO4: the insulating behavior—despite moderate Coulomb interactions usually present in 5d compounds—was even more intriguing, as the electronic and crystal structures are otherwise seemingly simple. The interplay of Coulomb correlations and spin–orbit coupling was indeed shown to be essential in driving the system insulation, leading to a state dubbed 'spin–orbit Mott insulator' [15, 16]. A flurry of further spin–orbit materials have by now been characterized, or known compounds have been reinvestigated in the light of the new insights. Iridium-based materials, where several families of compounds have been studied systematically, still hold a privileged position. Table 1 summarizes the structural, transport and magnetic properties of a selection of iridates. It is interesting to note that the large majority among them display insulating phases. The Ir4+ (5d5) state does not allow for a band insulating state without symmetry breaking, and magnetic order is an obvious candidate for helping to open the gap. Nevertheless, few compounds have been unambiguously characterized as Slater insulators.
Table 1. Main structural, transport and magnetic properties of Ir-based spin–orbit materials. In the third column, Ins. refers to insulator and MIT to metal–insulator transition. The notations AFM, FM and AIAO refer to an antiferromagnetic, ferromagnetic and all-in-all-out magnetic ordering, respectively.
| Iridium-based spin–orbit materials | |||||||
|---|---|---|---|---|---|---|---|
| Compound | Crystal struct. | Transport property | Magnetic ordering | Reference | |||
| CaIrO3 | Post-perovskite | Cmcm | Ins. | gap: 0.34 eV | AFM | K | [38–40] |
| NaIrO3 | Post-perovskite | Cmcm | Ins. | — | None | [41, 42] | |
| BaIrO3 | Monoclinic | C2/m | Ins. | gap: 0.05 eV | FM | K | [43–45] |
| SrIrO3 | Monoclinic | C2/c | Metal | None | [46–49] | ||
| α-Na2IrO3 | Honeycomb monoclinic | C2/c | Ins. | gap: 0.35 eV | Zig-zag AFM | K | [50–55] |
| α-Li2IrO3 | Honeycomb monoclinic | C2/c | Ins. | — | Spiral AFM | K | [56, 57] |
| β-Li2IrO3 | Hyperhoneycomb | Fddd | Ins. | — | Unconventional AFM | K | [58, 59] |
| γ-Li2IrO3 | Stripyhoneycomb | Cccm | Ins. | — | Unconventional AFM | K | [60] |
| Ba2IrO4 | K2NiF4-type | I4/mmm | Ins. | gap: 0.14 eV | AFM | K | [61–64] |
| Sr2IrO4 | Distorted K2NiF4-type | I41/acd | Ins. | gap 0.25 eV | Canted AFM | K | [15, 16, 21] |
| Ca4IrO6 | Hexagonal | Ins. | — | AFM | K | [65–67] | |
| Y2Ir2O7 | Pyrochlore | Ins. | — | AIAO | T = 155 K | [68, 69] | |
| Pr2Ir2O7 | Pyrochlore | Metal | None | [70, 71] | |||
| Nd2Ir2O7 | Pyrochlore | MIT | T = 36 K | AIAO | T = 36 K | [70, 72] | |
| Sm2Ir2O7 | Pyrochlore | MIT | T = 117 K | AIAO | T = 117 K | [70, 72] | |
| Eu2Ir2O7 | Pyrochlore | MIT | T = 120 K | AIAO | T = 120 K | [70, 73–75] | |
| Gd2Ir2O7 | Pyrochlore | Ins. | — | AIAO | T = 127 K | [70] | |
| Tb2Ir2O7 | Pyrochlore | Ins. | — | AIAO | T = 130 K | [70, 76] | |
| Dy2Ir2O7 | Pyrochlore | Ins. | — | AIAO | T = 134 K | [70] | |
| Ho2Ir2O7 | Pyrochlore | Ins. | — | AIAO | T = 141 K | [70] | |
| Er2Ir2O7 | Pyrochlore | Ins. | — | AIAO | T = 140 K | [76] | |
| Yb2Ir2O7 | Pyrochlore | Ins. | — | AIAO | T = 130 K | [77] | |
| Lu2Ir2O7 | Pyrochlore | Ins. | — | AIAO | T = 120 K | [78] | |
| Bi2Ir2O7 | Pyrochlore | Metal | None | [79, 80] | |||
| Sr3Ir2O7 | Monoclinic | C2/c | Ins. | gap: 0.1 eV | AFM | K | [46, 81–85] |
| Na4Ir3O8 | Hyperkagome | P4132 | Ins. | — | AFM | K | [86–88] |
| Ca5Ir3O12 | Hexagonal | P − 62m | Ins. | — | AFM | K | [66, 67, 89] |
| La2ZnIrO6 | Double-perovskite | P21/n | Ins. | — | FM | K | [90] |
| La2MgIrO6 | Double-perovskite | P21/n | Ins. | gap: 0.16 eV | AFM | K | [90, 91] |
| Pr2MgIrO6 | Double-perovskite | P21/n | Ins. | gap: 0.2 eV | AFM | K | [91, 92] |
| Nd2MgIrO6 | Double-perovskite | P21/n | Ins. | — | AFM | K | [92] |
| Sm2MgIrO6 | Double-perovskite | P21/n | Ins. | — | AFM | K | [92] |
| Eu2MgIrO6 | Double-perovskite | P21/n | Ins. | — | AFM | K | [92] |
| Gd2MgIrO6 | Double-perovskite | P21/n | Ins. | — | None | [92] | |
| Sr2CeIrO6 | Double perovskite | P21/n | Ins. | gap: 0.3 eV | AFM | K | [93–95] |
| Ba2YIrO6 | Double perovskite | Ins. | gap: 0.221 eV | None | [96] | ||
| Ba3IrTi2O9 | Hexagonal | P63mc | Ins. | — | None | [97, 98] | |
| Ba3ScIr2O9 | Hexagonal | P63/mmc | Ins. | — | None | [99] | |
| Ba3YIr2O9 | Hexagonal | P63/mmc | Ins. | — | FM | T = 4 K | [99] |
| Ba3ZnIr2O9 | Hexagonal | P63/mmc | Ins. | — | None | [100] | |


































Slightly more recently, attention focused on yet another class of 5d materials, namely osmium-based compounds. In this class fall, for example, ferroelectric LiOsO3 [30], as well as the prototypical Slater insulator NaOsO3 [31–36], where the loss of magnetic order with increasing temperature is accompanied by a closure of the insulating gap. It has been realized, however, that SOC can also have notable effects in 4d compounds, with prominent examples among ruthenium- and rhodium-based materials, where most interesting consequences for magnetic excitations have been discussed [37]. Table 2 gives an overview of the properties of a selection of osmates, ruthenates and rhodates. In the following discussion, we will restrict ourselves to the prototypical correlated iridate Sr2IrO4 and its 4d analog, Sr2RhO4.
Table 2. Main structural, transport and magnetic properties of Ru, Rh and Os-based spin–orbit materials. In the third column, Ins. refers to insulator and MIT to metal–insulator transition. The notations AFM, FM and AIAO refer to an antiferromagnetic, ferromagnetic and all-in-all-out magnetic ordering, respectively.
| Compound | Crystal struct. | Transport property | Magnetic ordering | Reference | |||
|---|---|---|---|---|---|---|---|
| Ruthenium-based spin–orbit materials | |||||||
| BaRuO3 | Cubic perovskite | Metal | FM | Tc = 60 K | [101–103] | ||
| CaRuO3 | Perovskite | Pnma | Metal | None | [104–106] | ||
| SrRuO3 | Perovskite | Pnma | Metal | FM | K | [104, 106, 107] | |
| Sr2RuO4 | K2NiF4-type | I4/mmm | Metal | None | [108, 109] | ||
| Ca2RuO4 | Distorted K2NiF4-type | Pbca or P21/c | MIT | T = 357 K | AFM | T = 110 K | [108, 110–112] |
| SrRu2O6 | Hexagonal | Ins. | — | AFM | K | [113, 114] | |
| Sr3Ru2O7 | Orthorhombic | BBcb | Metal | None | [106, 115] | ||
| Sr4Ru3O10 | Orthorhombic | Pbam | Metal | FM | K | [106, 116, 117] | |
| Rhodium-based spin–orbit materials | |||||||
| Li2RhO3 | Honeycomb | C2/m | Ins. | gap: 0.08 eV | None | [118, 119] | |
| Sr2RhO4 | Distorted K2NiF4-type | I41acd | Metal | None | [21, 120, 121] | ||
| Sr4RhO6 | Hexagonal | Ins. | gap: 0.1 eV | AFM | K | [122] | |
| Sr5Rh4O12 | P3c1 | Ins. | — | AFM | K | [66, 123] | |
| Osmium-based spin–orbit materials | |||||||
| BaOsO3 | Six-layer hexagonal 6H | Metal | None | [124, 125] | |||
| CaOsO3 | Perovskite | Pnma | Metal | None | [125] | ||
| SrOsO3 | Perovskite | Pnma | Metal | None | [125] | ||
| NaOsO3 | Perovskite | Pnma | MIT | T = 410 K | AFM | T = 410 K | [31–36] |
| Cd2Os2O7 | Pyrochlore | MIT | T = 226 K | AIAO | T = 226 K | [126–129] | |
| Ba2NaOsO6 | Double-perovskite | Ins. | — | FM | K | [130–132] | |
| Ba2LiOsO6 | Double-perovskite | Ins. | — | AFM | K | [131] | |
| Ba2CaOsO6 | Double-perovskite | Ins. | — | FM | K | [133, 134] | |
| Ba2YOsO6 | Double-perovskite | Ins. | — | AFM | K | [134, 135] | |


















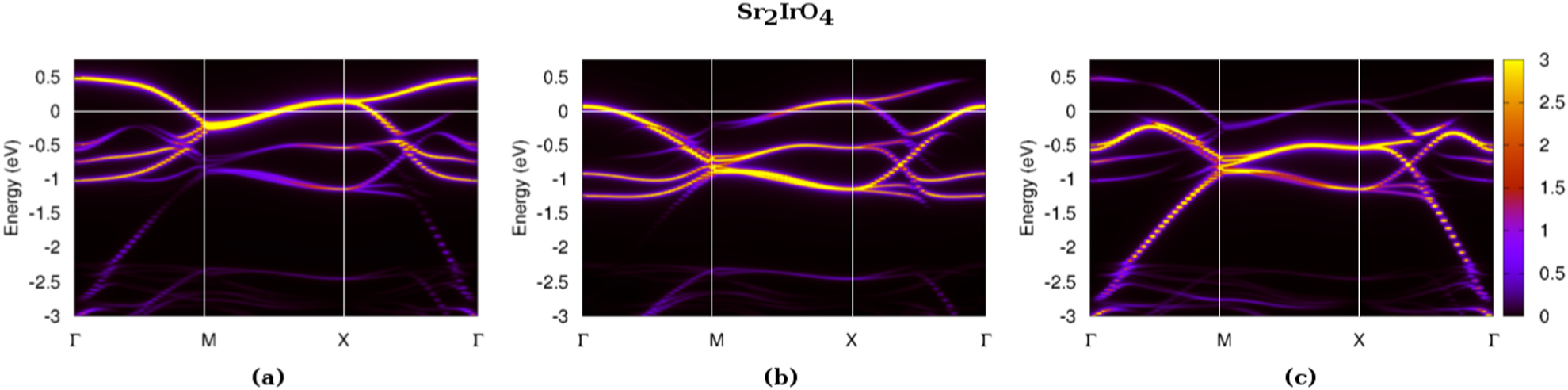
2.1. Correlated spin–orbit insulators: the example of Sr2IrO4
The 5d transition metal oxide (TMO) Sr2IrO4 has a tetragonal crystal structure, the symmetry of which is lowered from the K2NiF4-type, well-known in Sr2RuO4 or La2CuO4, by an rotation of its IrO6 octahedra around the -axis [136]. Each Ir atom accommodates 5 electrons and the standard picture neglecting spin–orbit interactions would give a '' ground state. However, this compound exhibits insulating behavior up to the highest measured temperatures, with a strongly temperature-dependent gap. The optical gap at room temperature is about 0.26 eV [137]. Below K, a canted-antiferromagnetic (AF) order sets in, with an effective local moment of 0.5 /Ir, and a saturation moment of 0.14 /Ir [138]. This phase has triggered much experimental and theoretical work [139–142], highlighting, in particular, the importance of the SOC.






Here, we focus on the paramagnetic phase, above 240 K, which is most interesting due to the persistence of the insulating nature despite the absence of magnetic order, as shown by transport measurements [15], by scanning tunneling microscopy and spectroscopy experiments [143], by angle-resolved spectroscopy [16, 144], time-resolved spectroscopy [145, 146] or optical conductivity [137].
Resonant inelastic x-ray spectroscopy (RIXS) experiments [15] proposed a picture early on in terms of states and states:




Since the quartet of states lies lower in energy than the doublet and the splitting between the and is large, neglecting any band dispersion would result in a configuration with one electron in the state. The DFT band structure displays a dispersion of width comparable to this splitting, leaving the question a priori open again. However, the bandwidth is narrowed due to structural distortions [21], and electronic correlations can then become effective and eventually drive the compound insulation.



Since the discovery of this mechanism, other Ir-based compounds (see table 1) have been classified as spin–orbit Mott insulators (Na2IrO3, pyrochlores, etc...). Recent theoretical studies also predict some fluoride material [147] to be in this class. The one-orbital nature of insulating Sr2IrO4 has contributed to intense activities attempting to dope the compound, with the hope of inducing a superconducting state as in the cuprates. Doping-induced metal–insulator transitions and the properties of the metallic phases have therefore become a hot topic, with studies of various compounds, e.g. Sr2IrO4 [144, 148], (Sr1−xLax)3Ir2O7 [149], Ca1−xSrxIrO3 [150], Ca1−xRuxIrO3 [151], Sr2Ir1−xRhxO4 [152, 153], Sr2Ir1−xRuxO4 [154], SrxLa11−xIr4O24 [155].
2.2. Correlated spin–orbit metals: the example of Sr2RhO4
It is natural that also in metallic 4d or 5d transition metal compounds, SOC can have notable consequences. An example of a 'spin–orbit correlated metal' is the end member SrIrO3 of the Ir-based Ruddlesden–Popper Srn+1IrnO3n+1 series [46] but also many Ru-,Rh- or Os-based transition metal oxides (TMOs) belong to this class (see tables 1 and 2). In these compounds, correlations are important enough to renormalize the Fermi surface-, albeit in a strongly spin–orbit coupling-dependent way. The respective roles of both effects have been worked out in some detail for several compounds, among which are SrIrO3 [46–48], Sr2RuO4/Ca2RuO4 [108, 109, 156] and Sr2RhO4 [19–21].
We will focus our attention in the following on Sr2RhO4, motivated by its structural proximity and isoelectronic nature to Sr2IrO4. Indeed, this TMO is the 4d counterpart of Sr2IrO4, both concerning structure and filling. To understand its Fermi surface requires the inclusion of both SOC and correlations [21]. It is composed of three pockets (see figure 8): a circular hole-like α-pocket around , a lens-shaped electron pocket and a square-shaped electron pocket with a mass enhancement of 3.0, 2.6 and 2.2, respectively [120].



In this review, we will put Sr2IrO4 and Sr2RhO4 in parallel, shedding light on the spectral properties of these compounds and elaborating on the notion of a reduced effective (spin–orbital) degeneracy that is crucial for their properties.
2.3. Spin–orbit coupling and cubic symmetry: the jeff picture
Necessary conditions for realizing a jeff picture are (1) a strong spin–orbit coupling constant and (2) an important cubic crystal field. These conditions are often met in crystalline structures where IrO6 octahedra are present (see table 1). Similar compounds based on Ru, Rh and Os also show such jeff states (see table 2). However, not all Ir-based structures belong to this case: we note that neither epitaxial thin films of IrO2 [157] nor the correlated metal IrO2 in its rutile structure [158, 159] exhibit such a state. We will now turn to a more precise description of that picture.

The spin–orbit interaction is one of the relativistic corrections to the Schrödinger–Pauli equation arising when taking the non-relativistic limit of Dirac's equation. It introduces a coupling between the spin and the motion—or, more precisely, the orbital momentum in the atomic case—of the electron. In a solid described within an independent-particle picture, spin–orbit coupling has the following general form:



where m0 is the electron mass, is the effective Kohn–Sham potential and denote the Pauli-spin matrices. Assuming that the potential close to the nucleus has spherical symmetry, the mean value of the spin–orbit interaction on the atomic state takes the more common form:




where , and denotes the mean value of the radial quantity in the state . Table 3 gives some values of the spin–orbit constant for 3d, 4d and 5d atoms. The SOC increases with the atomic number, explaining why spin–orbit materials are mostly found in 5d and 4d TMOs.





Table 3. Value of the spin–orbit constant in the d-valence shells of some transition metals. Data from Landolt–Börnstein database and [160] (3d), from [19, 161] (4d) and from [162] (5d).

| Atom | Z | (eV) | Atom | Z | (eV) | Atom | Z | (eV) |
|---|---|---|---|---|---|---|---|---|
| Fe | 26 | 0.050 | Ru | 44 | 0.161 | Os | 76 | 0.31 |
| Co | 27 | 0.061 | Rh | 45 | 0.191 | Ir | 77 | 0.40 |
| Cu | 29 | 0.103 | Ag | 47 | 0.227 | Au | 79 | 0.42 |



Due to the effect of SOC, a multiplet splitting arises in the d-orbitals. Figure 2 shows the multiplet splitting of d-orbitals due to the spin–orbit coupling as a function of the strength of a cubic crystal field .


Figure 2. Orbital diagrams for the d-shell of an atom as a function of the cubic crystal field and spin–orbit coupling , in a paramagnetic case. Starting from the d-shell in spherical symmetry, the cubic crystal field splits them into eg and t2g, while the SOC creates a six-fold J = 5/2 multiplet and a J = 3/2 quartet of lower energy. When both parameters are at stake, one gets a new multiplet structure where J remains a good quantum number but Jz does not. The initial J = 5/2 multiplet splits into a quartet and a doublet of lower energy, while the quartet J = 3/2 undergoes some redefinition inside its submanifold. The energetic splitting and the nature of the spin-orbitals depend on the ratio between . An exception is the doublet which is already of the form . In the limit where , as is the case in the compounds of our interest, one gets the celebrated splitting into eg, and .







Download figure:
Standard image High-resolution imageIn spherical symmetry the fine structure is composed of a six-fold J = 5/2 multiplet (in red) and a J = 3/2 quartet of lower energy (in blue), following 'Landé's interval rule'. The presence of a cubic crystal field splits further the six-fold multiplet. Indeed, the spin–orbit interaction in the cubic basis (eg and t2g in green and light green, respectively, in figure 2) can be reduced to two five-dimensional submatrices:

in the bases and , respectively. After diagonalization, the total angular momentum J remains a good quantum number, contrary to and one gets the following fine structure:



- a first quartet of J = 5/2 states (in red) with an energy
- a doublet of J = 5/2 states (in yellow) of energy
- a quartet of J = 3/2 states (in light blue) with an energy



In the limit of strong crystal field (), the J = 5/2 doublet (in yellow) remains invariant while the higher-energy quartet will tend to the usual eg states and the lower-energy J = 3/2 quartet will be composed of t2g states only, with an energy of .


Since the SOC matrix restricted to the t2g subspace is exactly the opposite of the SOC matrix of the p-states of a free atom, one usually labels these latter states by a jeff quantum number in analogy with the and multiplets, leading to the expressions given in equations (1) and (2). We point out that the doublet arises from the interplay of both cubic symmetry and SOC, whatever the strength of the crystal field. The corresponding eigenstates can indeed be written:




(where the right hand side is written using the J,mJ quantum numbers). This may explain the robustness of this doublet in spin–orbit compounds [163]. However, the splitting between the and multiplets follows the inverse Landé interval rule (with the above the states) only in the strong crystal field limit.




3. Interplay of spin–orbit interaction and Coulomb correlations from first principles
3.1. DFT+ DMFT calculations with spin–orbit coupling
Combined density functional theory (DFT) and dynamical mean-field theory (DMFT), as pioneered in [164, 165] (for a review, see [166, 167]), has made correlated electron systems accessible to first principles calculations. Over the years, various classes of systems ranging from transition metals [168–171], their oxides [11, 172–177], sulphides [178, 179], pnictides [4, 9, 180, 181], rare earths [182–184] and their compounds [185–187], including heavy fermions [188, 189], actinides [190, 191] and their compounds [192, 193] to organics [194], correlated semiconductors [195, 196], and correlated surfaces and interfaces [197–199] have been studied with great success. Besides intensive methodological developments (see e.g. [2, 3, 167, 200–203]), recent research activities continue to extend to new classes of materials. In this context, 4d and 5d oxides have also come into focus [21, 22, 62]. In this section, we review the technical aspects related to combined DFT+DMFT calculations in the presence of spin–orbit interactions. Since the applications we later focus on are 4d and 5d oxides in their paramagnetic phases, we restrict the discussion to this case.
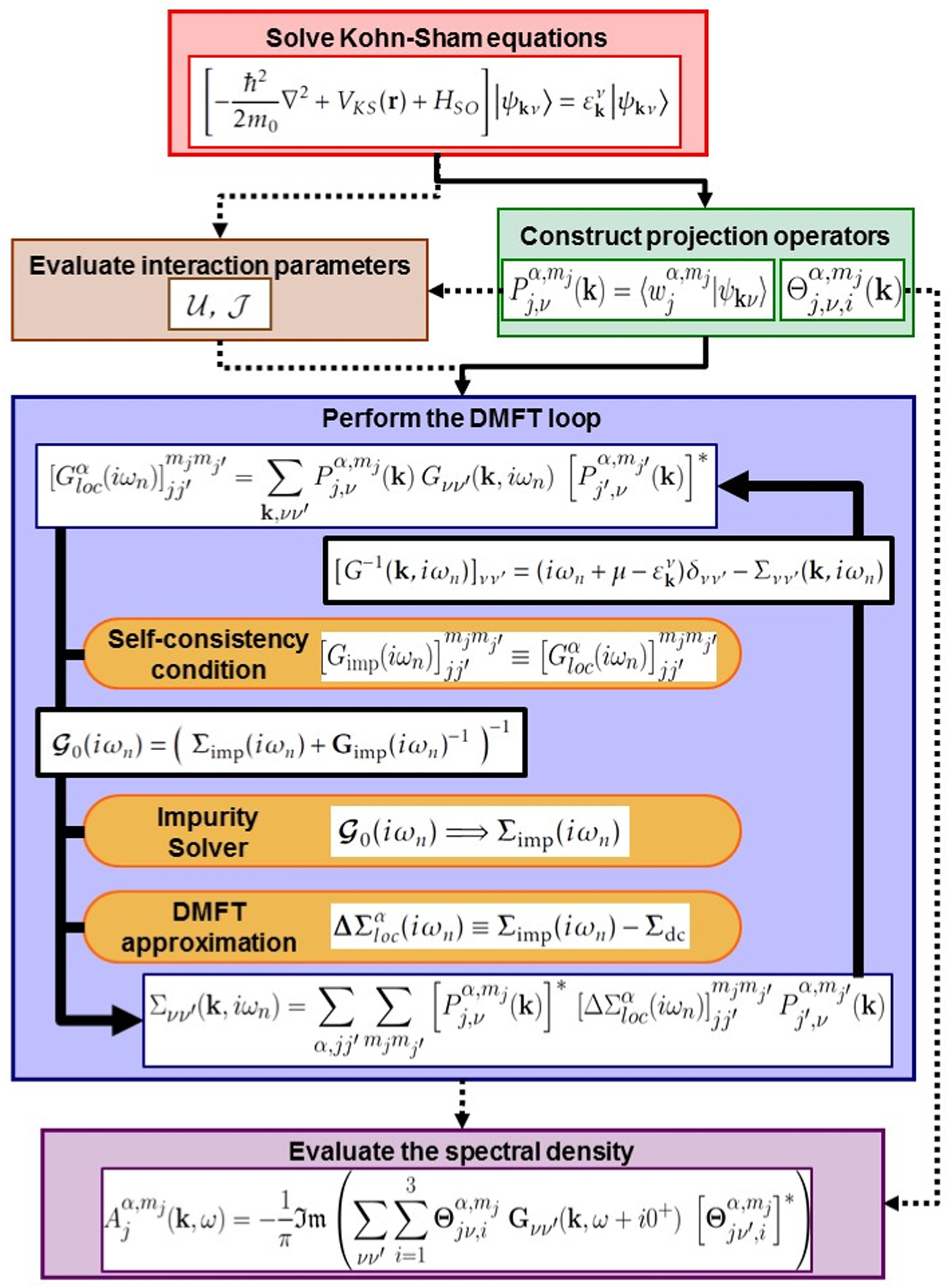
In DMFT, a local approximation is made to the many-body self-energy which can then be calculated from an effective atom problem, subject to a self-consistency condition (see figure 3).

Figure 3. Projector-based implementation of DFT+DMFT for calculations including spin–orbit coupling in the Kohn–Sham equations. Once the Kohn–Sham eigenstates are known, their projections to the correlated Wannier-like orbitals are calculated. One can then build an effective local many-body atomic problem, subject to a self-consistency condition, which is solved using an impurity solver: this defines the DMFT loop (see section 3.1). The interaction parameters can also be evaluated consistently using the projectors (see [204] and section 3.3). After convergence of the DMFT cycle, the chemical potential is updated and the spectral function can be evaluated using partial projectors (see appendix).





Download figure:
Standard image High-resolution imageThe notion of locality is understood in the sense of many-body theory as a site-diagonal form, with respect to atomic sites after representing the Hamiltonian in an atom-centered Wannier-type basis , where the index α labels the atom in the unit-cell, the angular momentum quantum numbers of the atomic orbital and σ the spin degree of freedom. Different choices are possible for the construction of the atom-centered orbitals, and the work reviewed here is based on the construction of projected atomic orbitals subject to a subsequent orthonormalization procedure [180].


The DMFT self-consistency cycle links the local effective atom problem to the electronic structure of the solid, via the transformation matrix from the Kohn–Sham states , labelled by their momentum their band index ν and their spin σ, to the resulting Wannier-like local orbitals . These key quantities are called projectors and denoted .




The main advantage of projector-based implementations of DFT+DMFT (see e.g. [180, 205, 206]) is that not only the DFT-based part of the calculations but also the determination of the local Green's function, used within the DMFT self-consistency condition, can be performed in any convenient basis set, and notably in the one used in the respective DFT code. Since the transformation of the DFT Hamiltonian matrix in that basis into the Kohn–Sham eigenset is known, it is sufficient to further determine the projections of the Kohn–Sham eigenstates onto the local orbitals used in the DMFT impurity problem. This is precisely the role of the projectors.


In [21], this construction was generalized to the case when spin is not a good quantum number anymore, and implemented within the framework of the DFT+DMFT implementation of [180]. Nowadays, it is available within the TRIQS/DFTTools package [207] that links the Wien2k code [208] to DMFT. We give here the main lines of this generalization of the projector-based DFT+DMFT formalism.
When taking into account SOC, the Kohn–Sham eigenstates are built out of both spin-up and spin-down states—in a similar fashion to the previously introduced and atomic states. Nevertheless, we can still write them in the following Bloch form:




where the index ν now runs over both spin and band indices. The state denotes the projection of the Kohn–Sham state onto its spin-σ contribution and is not an eigenstate of the Hamiltonian.

Using this decomposition, we can define the new projectors:

We define them in the standard complex basis, but allow for a basis transformation to quantum numbers j, mj (like and ) afterwards by means of a unitary matrix transformation in the correlated -space:




The main difference with the usual implementation where spin is a good quantum number is that there are now two projectors associated with each band index ν: with .


Using the decomposition (7) in the formulation of the self-consistency condition relating the lattice Green's function of the solid to the impurity model, the (inverse) Green's function of the solid is given by:

where are the (ν-dependent only) Kohn–Sham eigenvalues and is the approximation to the self-energy obtained by the solution of the DMFT impurity problem. It is obtained by 'mapping' the impurity self-energy to the local self-energy of the lattice and 'upfolding' it as:



with

Here, is the impurity self-energy, expressed in the local orbitals, and is the double-counting correction. Consequently, the equations of the DMFT loop (see figure 3) are formally the same as in the case without SOC, but the computations now involve matrices which are double in size.


The local Green's function is obtained by projecting the lattice Green's function to the set of correlated orbitals and summing over the full Brillouin zone,

In practice, the summation over momenta is done in the irreducible Brillouin zone only, supplemented by a standard symmetrization procedure, using Shubnikov magnetic point groups [209, 210].
The DMFT equations are solved iteratively: starting from an initial local Green's function (obtained from the 'pure' Kohn–Sham lattice Green's function using equation (13)), the Green's function of the effective environment in the impurity model is constructed. The impurity model is solved, allowing the evaluation of the local self-energy of the solid (see equation (11)) and a new lattice Green's function . The latter can then be projected again onto the correlated subset and the cycle is repeated until convergence is reached.



3.2. Computation of the Wannier projectors within the augmented plane wave framework
The present implementation is within a full-potential linearized augmented plane wave (FLAPW) framework, as realized in the Wien2k package [208]. With respect to the existing DFT+DMFT implementation [180] in this context, the main changes concern the projection technique for building the correlated orbitals: as discussed above, one has to take care of the fact that spin is no longer a good quantum number, leading to the more general construction of localized 'spin-orbitals'. The necessary modifications in the construction of the projectors are reviewed in the following.
As in the case without SOC, we still use the Kohn–Sham states within a chosen energy window to form the Wannier-like functions that are treated as correlated orbitals, and the construction of the Wannier projectors is done in two steps. First, auxiliary Wannier projectors are calculated—separately for each term—from the following expression:




A description of the augmented plane wave (APW) basis can be found in [180]. We use the same notations e.g. for the coefficients and the overlap matrix as introduced there.


One performs an orthonormalization step in order to get the Wannier projectors . The overlap matrix between the correlated orbitals is defined by:

![$\left[O\left(\mathbf{k}\right)\right]_{(m\sigma ),\left({{m}^{\prime}}{{\sigma}^{\prime}}\right)}^{\alpha,{{\alpha}^{\prime}}}$](https://content.cld.iop.org/journals/0953-8984/29/26/263001/revision2/cmaa648fieqn140.gif)


leading to the final projectors:

which are then further transformed into a j,mj basis as described above (see equation (9)).
3.3. Effective local Coulomb interactions from first principles
Hubbard interactions U—obtained as the static () limit of the on-site matrix element within the 'constrained random phase approximation' (cRPA)—have by now been obtained for a variety of systems, ranging from transition metals [211] to oxides [204, 212–215], pnictides [181, 186, 216, 217], f-electron elements [218] and compounds [187], to surface systems [219], and several implementations within different electronic structure codes and basis sets have been done, e.g. within linearized muffin tin orbitals [211, 220], maximally localized Wannier functions [212, 216, 221] (as elaborated in [222]), or localized orbitals constructed from projected atomic orbitals [204]. The implementation into the framework of the Wien2k package [204] made it possible for Hubbard U's be calculated for the same orbitals as the ones used in subsequent DFT+DMFT calculations, and, to our knowledge, [21] was indeed the first work using in this way consistently calculated Hubbard interactions in a DFT+DMFT calculation. Systematic calculations investigating the basis set dependence for a series of correlated transition metal oxides revealed further interesting trends, depending on the choice of the low-energy subspace. In contrast to common belief until then, Hubbard interactions increase, for example, with the principal quantum number when low-energy effective models encompassing only the t2g orbitals are employed. These trends can be rationalized by two counteracting mechanisms, the increasing extension of the orbitals with increasing principal quantum number and the less efficient screening by oxygen states [204]. We will come back to this point below, in the context of the cRPA calculations for our target compounds.


In the following, we review the specificities involved when determining the Hubbard interactions for our target spin–orbit compounds. We hereby use the same notations as in [204].
We start from the standard Hubbard–Kanamori Hamiltonian Hint which allows us to describe the interactions between t2g orbitals within a Hamiltonian restricted to the t2g-space:

where is the intra-orbital Coulomb repulsion term and ( with cubic symmetry) the inter-orbital Coulomb interaction which is reduced by Hund's exchange . (m and n run over the three t2g orbitals and σ stands for the spin).




To draw the link between the cRPA calculations and this model Hamiltonian, the terms , and are understood as the Slater-symmetrized effective interactions in the t2g subspace, related to the Slater integrals F0, F2 and F4 as:




The last relation is redundant since .


One now transforms Hint into the basis using the unitary matrix transformation . Keeping only density-density terms, Hint becomes:



Here, the index j is a shortcut notation for the quantum number and . The reduced interaction matrix has the following form:




We use the standard convention that denotes −mj, as is usually done for spin degree of freedom. The ordering of the orbitals is: , and blocks are emphasized to ease the reading of the matrices.





3.4. Technicalities of the DMFT calculation
For the solution of the quantum impurity problem we apply the continuous-time quantum Monte Carlo method (CTQMC) in the strong-coupling formulation [223]. We are able to perform calculations at room temperature ( eV−1) with reasonable numerical effort. In our calculations, we typically use around Monte Carlo sweeps and 28 k-points in the irreducible Brillouin zone.


Since the CTQMC solver computes the Green's function on the imaginary-time axis, an analytic continuation is needed in order to obtain results on the real-frequency axis. A continuation of the impurity self-energy using a stochastic version of the maximum entropy method [224] yields real and imaginary parts of the retarded self-energy. From those, we calculate the momentum-resolved spectral function using partial projectors introduced in the appendix.

During the calculations we use the fully localized limit (FLL) expression for the double-counting:

where j and run over the states and Nc is the total occupancy of the orbitals. (Since each orbital is doubly degenerate in mj, Nc/2 is used in the term containing J). Moreover, we neglect the off-diagonal terms in the local Green's functions (particularly, we neglect the term between the and the which we checked to be two orders of magnitude smaller than the diagonal terms, in the chosen basis).





4. Electronic structure of Sr2IrO4 and Sr2RhO4
4.1. Electronic structure of Sr2IrO4 and Sr2RhO4 within DFT-LDA
The Kohn–Sham band structures of Sr2IrO4 and Sr2RhO4 within the local density approximation and in the presence of spin–orbit coupling (LDA + SO) are represented in figures 4(d) and (e). For Sr2IrO4, we use the lattice parameters measured at 295 K in [225], and for Sr2RhO4 those measured at 300 K in [226].

Figure 4. Kohn–Sham band structures within LDA + SO of Sr2RhO4 and Sr2IrO4 assuming that they crystallize without distortions in a K2NiF4 structure ((a)–(b)), of Sr2IrO4 in a supercell containing four 'undistorted' unit-cells (c) and of 'real' Sr2RhO4 and Sr2IrO4 ((d)–(e)). The reduction of the first Brillouin zone, when the crystal symmetry is lowered, is also shown. The eg states are plotted in yellow () and red () while the O-2p states are in black. In the t2g manifold, the are plotted in purple, the mj = 3/2 in light blue and the mj = 1/2 in green.





Download figure:
Standard image High-resolution imageThe LDA + SO band structures for Sr2IrO4 and Sr2RhO4 are very similar, as a consequence of both the structural similarity and the key role of spin–orbit coupling in these compounds. The eg-states ( in red and in yellow) start at about 1–1.5 eV, and are fully separated from the t2g-manifold which lies around the Fermi level and overlaps at lower energies with the oxygen 2p-states (black). Given the filling and the four-atom unit cell of both compounds, a metallic solution is obtained within LDA for both Sr2RhO4 and Sr2IrO4—at variance with experiments for Sr2IrO4. Among the t2g-manifold (in green), only the four highest-lying bands, highlighted in blue, cross the Fermi level: this is suggestive of the existence of a separated half-filled -derived band, which—within a four-atom unit cell—corresponds to a quartet of bands at each k-point. We stress, however, that the true picture is much more subtle: in fact, and overlap (see the band structure between the and the M-point for instance) and the identification of the upper four bands as the states is too simplistic. We will come back to this point below.








To get a better understanding of the Kohn–Sham band structures of Sr2RhO4 and Sr2IrO4, we study artificial compounds where both the structural distortions and the spin–orbit coupling have been switched off. Figures 5(b) and (c) depict the LDA band structure of such 'idealized undistorted Sr2RhO4 and Sr2IrO4'. Neglecting the rotation of about of their IrO6 and RhO6 octahedra around the c-axis leads to a K2NiF4-type crystal structure, like in Sr2RuO4, the well-known LDA band structure of which is plotted in figure 5(a).


Figure 5. Kohn–Sham band structures of Sr2RuO4 (a), Sr2RhO4 (b) and Sr2IrO4 (c) within LDA (and without spin–orbit coupling), artificially assuming that both Sr2RhO4 and Sr2IrO4 crystallize in the same K2NiF4 structure as their Ru-counterpart. For Sr2RuO4, we use the lattice parameters at 300 K given in [227]. The t2g-dominated bands are plotted in green (dxy) and blue (dxz and dyz) while the eg bands are in red () and yellow (), and the O-2p states are plotted in black.


Download figure:
Standard image High-resolution imageThe similarity of the three band structures is obvious. Around the Fermi level, one distinguishes the three t2g bands. The dxy-band (green) reaches out to lower energies and overlaps with the oxygen 2p-states (black). The eg-states ( in red and yellow), higher in energy, cut the Fermi level in both Sr2IrO4 and Sr2RhO4 due the additional electron remaining in the d-manifold, contrary to Sr2RuO4, which has actually a mere -filling. The larger extension of the 5d orbitals (see figure 1) explains the wider bandwidth observed for Sr2IrO4 in comparison to Sr2RhO4: the dxy band reaches the value of −3.5 eV in , while it remains above −3 eV for the 4d counterparts. Another consequence of this wider extension is the stronger hybridization between the 5d states with the oxygen p-states, which are located 1 eV lower in energy in Sr2IrO4 than in the 4d-TMOs.




Re-introducing the effects of the spin–orbit coupling in Sr2RhO4 and Sr2IrO4 (but without considering the structural distortions) modifies these Kohn–Sham band structures to those shown in figures 4(a) and (b). The t2g bands are the most affected, while the eg bands are slightly shifted as a consequence of the topological change in the t2g manifold. A detailed study of the character of these band structures confirms the decoupling between eg and t2g states (see also [19, 20]). The cubic crystal field at stake in these compounds is indeed much larger than the energy scale associated with the spin–orbit coupling of about eV and eV for Sr2IrO4 and Sr2RhO4 respectively.


The jeff picture is thus justified in both Sr2IrO4 and Sr2RhO4: the t2g orbitals split into a quartet of states and a higher lying doublet (see figure 2). Each state is doubly degenerate in , since we observe the system in its paramagnetic phase at room temperature and the crystal structure has a center of inversion. Therefore we still refer to them as the ' band' and the two ' bands' in the following. The three bands can easily be identified: the one (light green) lies above the two ones (mj = 3/2 in light blue and mj = 1/2 in violet). The three jeff bands are well-separated all along the -path, and more generally in the whole Brillouin zone. Since the spin–orbit coupling is half the size in Sr2RhO4, the splitting between the jeff bands is reduced by a factor of 2, as one can see, for instance, at X or .










To draw the link between the 'undistorted' band structures and the realistic ones, we plot in figure 4(c) the LDA + SO band structure of the undistorted Sr2IrO4 in a supercell containing four unit cells. Each band is now folded four times and we provide a scheme of the two first Brillouin zones in the plane to understand the correspondence between the high-symmetry points of each structure.

Comparing figures 4(c) and (e) highlights the key role of the structural distortion in Sr2IrO4: a hybridisation between two neighboring Ir dxy and orbitals via the in-plane oxygens is now allowed and pushes the t2g and eg bands apart. Another consequence of the distortions is the general narrowing of the jeff bandwidth, which is of crucial importance in driving the compound insulation, as we will see below.

Finally, comparing figures 4(c) and (e) gives more insight into the nature of the four highest-lying bands (blue) of figure 4(e). Along the M − −X direction, each quartet of the jeff bands remain well-separated, and overlap in the other direction and M − X. As a result, the bands cross the Fermi-level closest to the -point, while the other crossings are due to the bands. The identification of the upper four bands in Sr2IrO4 as 'pure' states is thus too simplistic, implying the need for a Hamiltonian containing more than one orbital in a realistic calculation.







The same mechanisms are important in Sr2RhO4 even though we do not display the orbital characters here: the four highest-lying bands, highlighted in blue in figure 4(d) exhibit a mixed character of type and . Moreover, thanks to the distortions which allow the opening of a gap between t2g and eg bands, the LDA + SO Fermi surface becomes qualitatively similar to the experimental one; as shown in figure 8(a), they both contain three closed contours: a circular hole-like α-pocket around , a lens-shaped electron pocket and a square-shaped electron pockets . However, the striking discrepancies in the size of the pockets point out a subtle deficiency in the LDA for Sr2RhO4 [19, 20].





4.2. Wannier functions
We have derived the Wannier functions associated with the manifold for both Sr2IrO4 and Sr2RhO4, using the framework introduced in section 3.1. Because of the mixed character of the four bands that cross the Fermi level in Sr2IrO4 and Sr2RhO4, the local effective atomic problem used in the DMFT cycle must contain the three jeff orbitals and thus accommodate five electrons. We construct Wannier functions for the jeff orbitals from the LDA + SO band structure of Sr2IrO4 and Sr2RhO4, using an energy window [−3.0,0.5] eV for Sr2IrO4 and an energy window [−2.67;0.37] eV for Sr2RhO4.

Figures 6 and 7 depict the projection of these Wannier functions on the LDA + SO band structure. The similarities between figures 6 and 4(c) are numerous, thus confirming our previous band character analysis. Table 5 gives the decomposition of these local Wannier functions on the t2g manifold and their respective occupation.

Figure 6. LDA + SO band structure of Sr2IrO4, projected on the (a), (b), and (c) spin-orbitals.





Download figure:
Standard image High-resolution image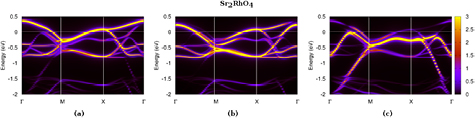
Figure 7. LDA + SO band structure of Sr2RhO4, projected on the (a), (b), and (c) spin-orbitals.





Download figure:
Standard image High-resolution imageTo obtain deeper insights into the nature of these Wannier orbitals, table 4 gives the coefficients of the local Wannier orbitals obtained from the LDA + SO band structure of 'undistorted' Sr2IrO4 using an energy window [−3.5, 0.8] eV. The results agree well with the standard jeff picture (see equations (1) and (2)) in both modulus and phase. Discrepancies are mostly due to the elongation of the IrO6 along the c-axis, which introduces an additional tetragonal field between the t2g states. This effect also explains the lifting of the degeneracy of the two ( and ) states and implies the reason why the is slightly more than half-filled.




Table 4. Coefficients and occupation (within LDA + SO) of the jeff Wannier orbitals in 'undistorted' Sr2IrO4. The discrepancy between these coefficients and those given in equations (1) and (2) are due to the small elongation of the octahedra along the c-axis.
| 'Undistorted' Sr2IrO4 | |||
|---|---|---|---|
| dxy | ±0.6605 | +0.7508 | 0 |
| dxz | ±0.5309 i | −0.4670 i | −0.7071 i |
| dyz | +0.5309 | 0.4670 | 0.7071 |
| Occupation (LDA + SO) | 1.20 | 1.92 | 1.86 |











Because of the hybridization between the dxy and orbitals in the distorted structures, we had to define in practice 'effective and states', which remain close to the atomic jeff picture but take into account a small amount of character (see table 5). The coefficients have been calculated such that the density matrix of the local atomic problem is the closest possible to the diagonal form4. In addition to the hybridization, the construction of the 'effective jeff' also takes into account the tetragonal crystal field due to the elongation of the octahedra in each crystal structure; this explains the discrepancies with the standard coefficients given in equations (1) and (2). We note that the coefficients obtained for the state of Sr2IrO4 are equivalent to those obtained in the AF phase in [139].






Table 5. Modulus of the coefficients of the jeff Wannier orbitals in Sr2IrO4 and Sr2RhO4. The occupation within LDA + SO and the charge within LDA + SO+DMFT of each atomic Wannier orbital are also provided, showing how electronic correlations enhance the spin–orbital polarization.
| Sr2IrO4 | Sr2RhO4 | |||||
|---|---|---|---|---|---|---|
| Wannier orbitals | ||||||
| 0.0388 | 0.0766 | 0 | 0.0100 | 0.0302 | 0 | |
| d xy | 0.4499 | 0.8889 | 0 | 0.3153 | 0.9485 | 0 |
| d xz | 0.6309 | 0.3193 | 0.7071 | 0.6710 | 0.2231 | 0.7071 |
| d yz | 0.6309 | 0.3193 | 0.7071 | 0.6710 | 0.2231 | 0.7071 |
| occupation (LDA + SO) | 1.16 | 1.98 | 1.84 | 1.42 | 1.96 | 1.64 |
| charge (LDA + SO+DMFT) | 1.02 | 2.00 | 1.98 | 1.26 | 1.98 | 1.76 |















Finally, comparing the occupation of the orbitals in tables 4 and 5 highlights again the role of the hybridisation between the dxy and orbitals which pushes the band further below the Fermi level close to . As a result, the four bands that cross the Fermi level are formed only by the and orbitals and the tend to be close to half-filling. Similar conclusions were drawn for the AF phase within a variational cluster approximation (VCA) approach in [140]. Similar conclusions hold for Sr2RhO4.








4.3. Effective Hubbard interactions from cRPA
After defining the jeff Wannier orbitals, we evaluate the local Coulomb interaction in the effective atomic problem within cRPA [204, 211], as explained in section 3.3. For reasons of computational resources, the cRPA calculations were performed in the case without distortions (without the rotations of the octahedra, hence considering only one formula-unit in a unit-cell) and without SOC. To mimic the effect of the distortions, the eg states are shifted up to their energetic position in the presence of distortions. We find eV and eV for Sr2IrO4 and eV and eV for Sr2RhO4. These parameters lead to the following local interaction matrices for Sr2IrO4:






and for Sr2RhO4:


where the values are in eV and the ordering of the orbitals is: , , and denotes −mj. We remind the reader that and . Since we have used 'effective jeff' Wannier orbitals instead of the standard definition given in equations (1) and (2), some discrepancies with the formulae given in equation (20) and in [22] can be observed. Contrary to common belief, the Hubbard interactions are smaller in the 4d-TMO than in its 5d-counterpart. This might seem counterintuitive at first sight, since the 5d-orbitals are more extended than the 4d ones, but finds its explanation in more efficient screening in the 4d material: As shown in figures 4(d) and (e), the hybridization between the Rh-4d states and the O-2p is weaker in Sr2RhO4 than in Sr2IrO4. Correspondingly, the energetic position of the O-2p bands is closer to the Fermi level by about 1 eV, and as a result, the Coulomb interactions are screened more efficiently in Sr2RhO4 than in Sr2IrO4, explaining the observed trend.







4.4. Correlated electronic structure of Sr2IrO4 and Sr2RhO4
DFT+DMFT calculations following the procedure described in section 3.1 indeed find an insulating solution for Sr2IrO4 and a correlated metal for Sr2RhO4 [21], in agreement with experiment. The difference in the metallic versus insulating nature of Sr2RhO4 and Sr2IrO4 can be traced back to the different spin–orbital polarization in the three jeff orbitals, which is enhanced by Coulomb correlations.
The occupations of the jeff Wannier orbitals within LDA + SO and LDA + SO+DMFT are provided in table 5. In Sr2IrO4, one detects a considerable spin–orbital polarization already at the LDA + SO level: the four states are almost filled with and while the states thus slightly exceed half-filling with n1/2 = 1.16 (as in the 'ideal undistorted' case). Taking into account Coulomb correlations within DMFT opens a gap of about 0.26 eV [21] and enhances the spin–orbital polarization, such as to fill the states entirely, leading to a half-filled state. This is thus the celebrated ' -picture' [16], which comes out here as a result of the calculations, rather than being an input as in most model Hamiltonian calculations.







A different picture emerges for Sr2RhO4 according to table 5: while the spin–orbital occupations display some polarization at the LDA + SO level, the smaller SOC—and thus the smaller effective splitting between the jeff bands—leads to a picture where only the state is entirely filled, while both and live at the Fermi level. This spin–orbital polarization is enhanced by Coulomb correlations—just as in Sr2IrO4—but this enhancement is not enough to fill both states entirely and obtain a half-filled state. The higher effective degeneracy, together with the smaller value of , eventually leaves Sr2RhO4 metallic.








4.5. Spectral properties of Sr2RhO4: theory vs. experiment
We now turn to the calculated spectral function of the spin–orbital correlated metal Sr2RhO4 that we analyze in comparison to experiment.
Figure 8 depicts the Fermi surface of Sr2RhO4 within LDA + SO (left panel) and LDA + SO+DMFT (right panel) in the plane, on which we superimpose the experimental measurement from [120]. Table 6 gives more quantitative insight to ease the comparison between the different topologies. All three Fermi surfaces, the two theoretical ones and the experimental one, are qualitatively similar with three closed contours: a circular hole-like α-pocket around , a lens-shaped electron pocket and a square-shaped electron pockets . These two structures merge in the undistorted tetragonal zone (dashed blue line in figure 8) to a large electron-like pocket β.





Figure 8. Calculated Fermi surface of Sr2RhO4 in the plane within LDA + SO (left panel) and LDA + SO+DMFT (right panel). Superimposed is the experimentally measured Fermi surface. Adapted with permission from [120]. Copyright 2006 by the American Physical Society.

Download figure:
Standard image High-resolution imageTable 6. Comparison of the Fermi surface (FS) parameters evaluated within LDA + SO, within LDA + SO+DMFT and ARPES [120]. For each α, and pocket, the FS volume A is defined as a percentage of the two-dimensional BZ volume (using the experimental lattice parameters (a = 5.45 Å)). The Fermi velocity is obtained from the slope of the band dispersion at the Fermi level. The cyclotron mass is calculated using the same method as described in [120]: .





| α | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| LDA | DMFT | Exp. | LDA | DMFT | Exp. | LDA | DMFT | Exp. | |
| FS volume A (% BZ) | 18.4 | 10.1 | 6.1(4) | 4.5 | 6.2 | 8.1(5) | 10.0 | 7.6 | 7.4(4) |
| (eV · Å) | 1.252 | 0.645 | 0.41(4) | 1.260 | 0.674 | 0.55(6) | 1.260 | 0.674 | 0.61(6) |
| m* (me) | 1.70 | 2.44 | 3.0(3) | 0.83 | 1.83 | 2.6(3) | 1.24 | 2.02 | 2.2(2) |



Comparing figures 8(a) and (b) highlights the key role of electronic correlations: they decrease the radius of the α pocket from 0.26–0.29 to 0.21 and decrease the radius of the large β pocket from 0.69–0.72 to 0.67–0.70 , thus enlarging the and pockets such that their volumes are well-reproduced within LDA + SO+DMFT (see table 6). As a result, the agreement between LDA + SO+DMFT data and the experimental measurements becomes quantitatively excellent.






To go further in the analysis, figure 9 depicts the momentum-resolved spectral function, as well as its orbital-resolved version. The completely filled state is visible (panel (d)), as well as the partially filled character of the (panel (c)) and states (panel (b)). A detailed comparison with angle-resolved photoemission data from [121] (blue dashed line on the figure) shows that the band dispersion around the Fermi level is well-reproduced, while some discrepancies are observed for the structures experimentally observed along and at lower energy. These features, reminiscent of the bands, are indeed about 0.05 eV higher in energy in our calculated spectral function.










Figure 9. Calculated momentum-resolved spectral function of Sr2RhO4 within LDA+DMFT (a) and its orbital-resolved versions for the states (b), the (c) and the completely-filled (d). The dashed blue line on panel (a) is the reproduction of the ARPES structure from [121].





Download figure:
Standard image High-resolution imageFrom figures 9(b) and (c), one observes that the Fermi level is crossed by the renormalized band at 0.20 along and at 0.21 along , while the renormalized band is responsible for all other crossings. This allows us to label the hole-like α-pocket as being of type, whereas the two other pockets and are mostly of type . Using the quasiparticle weight of each state (Z1/2 = 0.535 and ), we evaluate the Fermi velocity at each crossing along the path : we find a huge variation in the values depending on and give in table 6 their mean value over the Brillouin zone. Finally, using the same method as described in [120], we evaluate the cyclotron mass based on the approximate formula used there: . The DMFT results shown in table 6 show a substantial improvement over DFT when compared to experiments.













![$\left[\Gamma MX \Gamma \right]$](https://content.cld.iop.org/journals/0953-8984/29/26/263001/revision2/cmaa648fieqn346.gif)



5. The effective orbital degeneracy as a key quantity determining the correlation strength
In section 4.4, we have identified the spin–orbital polarization as a key factor in explaining the different behavior of Sr2RhO4 and Sr2IrO4.
In Sr2IrO4, Coulomb correlations enhance the spin–orbital polarization, such as to fill the states entirely, leading to a half-filled one-band picture, while in Sr2RhO4 the final situation is an effective two-orbital system containing three electrons. This situation is akin to correlation-induced enhancements of orbital polarization also observed in other transition metal oxides. In the distorted 3d1 perovskites LaTiO3 and YTiO3, for example, it was argued [10] that the interplay of structural distortions and Coulomb correlations leads to a suppression of orbital fluctuations in the t2g-manifold, favoring a particular orbital composition selected by crystal and ligand field effects. At the LDA level, 0.45 [0.88] electrons are found in this particular orbital in LaTiO3 [YTiO3], while Coulomb correlations as described by LDA+DMFT lead to an occupation of 0.88 [0.96] electrons.


In these systems, this reduction of effective orbital degeneracy was shown to be key to their insulating nature since the critical interaction strength needed to localize the single electron is thus effectively determined by the one of a single-orbital system, instead of the one of a three-fold degenerate t2g-manifold. Within DMFT, the critical Hubbard interaction scales with the square-root of the orbital degeneracy N for the lower critical interaction of the phase coexistence region of the first order Mott transition, while the upper critical interaction varies with N [228].
Localizing electrons in a single-orbital system therefore needs a critical interaction which is smaller by a factor of roughly 3 as compared to the degenerate case. This was demonstrated to be crucial for the difference in behaviors in the series of d1 compounds SrVO3, CaVO3, LaTiO3, YTiO3, where the former are three-fold degenerate metallic systems, whereas the latter realize the single-orbital Mott state.
The situation in the iridates is analogous with the purely formal difference that one is dealing with a one-hole situation instead of one electron. Furthermore, the strong spin–orbit interaction is instrumental in the suppression of the degeneracy, which is the net result of structural distortions, spin–orbit coupling and Coulomb correlations.
This discussion highlights an important aspect of the physics of transition metal oxides, often neglected when considering band filling and interaction strength only: the effective orbital degeneracy is a crucial tuning parameter for electronic behavior, suggesting that the popular picture distinguishing filling-controlled and bandwidth-controlled Mott transitions [1] should be complemented by a 'third axis' and the notion of degeneracy-controlled Mott behavior.
Crystal and ligand fields, together with spin–orbit coupling and the Coulomb correlations themselves, are the driving forces for establishing a given effective degeneracy. At the level of the calculations, this effective degeneracy is both an outcome of the calculation and a determining factor of the properties of the given compound.
6. Conclusions and perspectives
The common belief about electronic Coulomb correlations being less important in 4d and 5d compounds as compared to 3d transition metal oxides, was overruled by insights into the role of spin–orbit coupling in the insulating behavior of iridates [16] and for the Fermi surface topology of Sr2RhO4 [19, 20].
Here, we have reviewed recent work on a first principles many-body description of such effects within a dynamical mean-field framework. We have highlighted the notion of the effective degeneracy of the system as a crucial parameter determining the physical properties of a system. The effective degeneracy is the result of a complex interplay of structural distortions, spin–orbit coupling and Coulomb correlations. We have stressed the analogy of the Mott insulating picture for Sr2IrO4 with the insulating nature of LaTiO3 and YTiO3 in the 'degeneracy-controlled Mott transition' series of d1 perovskites (SrVO3, CaVO3, LaTiO3, YTiO3) [10].

In Sr2IrO4 and Sr2RhO4 the difference in degeneracy is itself a consequence of the quantitative aspects of the physics of these two compounds: all three decisive elements—structural distortions, spin–orbit coupling and Hubbard interaction—are smaller in Sr2RhO4 than in Sr2IrO4 and this quantitative difference in the electronic parameters translates into a qualitative difference in the resulting properties.
We have analyzed in detail the spectral properties of Sr2RhO4, a spin–orbit correlated 4d metal where the effective degeneracy is reduced by spin–orbit coupling and correlations but not to the point such as to induce a Mott insulator. The calculated spectral properties and Fermi surface are in excellent agreement with experimental data. A detailed analysis of the spectral properties of Sr2IrO4 is left for future work.

Acknowledgments
This work was supported by the ERC Consolidator Grant CORRELMAT (grant 617196), the French ANR under project IRIDATES, and IDRIS/GENCI under project t20169313. M.A. is supported by a START program of the Austrian Science Fund (FWF), grant number Y746.
Appendix. Generalized partial Θ-projectors and spectral function
In order to calculate quantities for a given atom α and a particular orbital (spin) character j (mj)—such as the spectral functions —a set of partial projectors called ' -projectors' was built. Contrary to the previously introduced Wannier projectors , their definition is not restricted to the correlated orbitals only. The formalism of these partial projectors was initially introduced in [180] and was extended to the case where spin is not a good quantum number anymore, in [21].



Inside the muffin-tin sphere associated with an atom α, one can write the spin-σ contribution of the eigenstate as:


where the basis is not orthonormalized, as already mentioned in [180]. That is why, to make the calculations easier, one introduces an orthonormal basis set for each atomic orbital . These orbitals are defined from the initial basis as follows:






We can then rewrite equation (A.1) as:

The matrix elements are the ' -projectors', which are thus defined by:



Contrary to the implementation of [180], there are now a couple of -projectors associated with each band index ν, with , since spin is not a good quantum number anymore.



We have introduced here the -projectors in the complex spherical harmonics basis. As for the Wannier projectors, it is of course possible to get the -projectors in any desired j,mj basis:



Finally, the spectral function , which is defined by:


is obtained for a given atom α with orbital character (j,mj) through the following formula:

where the band indices ν, run over both spin and orbital quantum number.

Footnotes
- 4
With the obtained coefficients, the off-diagonal terms remaining in the local Green's functions between the and are smaller than 0.05. In practice, the coefficients were chosen to be real. This can be done in the local problem since only density-density terms were kept for the interaction terms and off-diagonal terms of the density matrix were neglected.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}