


Abstract
In this paper a computational high-throughput screening (HTS) approach to the search for alternative permanent magnetic materials is presented. Systems considered for a start are binary intermetallic compounds composed of rare-earth (RE) and transition metal (TM) elements. With the tight-binding-linear muffin-tin-orbital-atomic-sphere-approximation (TB-LMTO-ASA) method of density functional theory (DFT) a variety of RE–TM intermetallic phases is investigated and their magnetic properties are obtained at rather low computational costs. Next, interstitial elements such as boron, carbon and nitrogen in these phases are considered. For promising candidate phases with high and stable spontaneous ferromagnetic polarization, the calculated local magnetic moments and exchange coupling parameters, as obtained from TB-LMTO-ASA calculations, are then used for Monte Carlo simulations to identify candidates with sufficiently high Curie temperatures (Tc). Finally, magnetocrystalline anisotropy constants (K1) of the most promising candidate phases are calculated with accurate, potential-shape-unrestricted DFT calculations using the Vienna ab initio simulation package. The computational HTS procedure is illustrated by results for a selection of hard-magnetic RE–TM phases like RETM5, RE2TM17 and RE2TM14B.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
GENERAL SCIENTIFIC SUMMARY Introduction and background. In the recent years of rapid technological development for electromobility and renewable energy, permanent magnets based on rare-earth (RE) and transition-metal (TM) elements have become of major importance due to their multiple applications in motors, sensors, etc. Due to the limited resources of RE elements and a latent supply monopole on the market, the ultimate goal of computational materials science is to discover new magnetic materials with the lowest possible RE content and improved performance.
Main results. We developed a computational high-throughput screening (HTS) approach to the search for alternative magnetic materials. Systems considered for a start are binary intermetallic compounds composed of RE and TM elements. Using fast computation based on density functional theory (DFT), a variety of RE–TM phases are investigated and their magnetic properties obtained at low computational costs. For the materials selected as promising, it is possible to compute the Curie temperature and magnetocrystalline anisotropy constants with accurate DFT methods and thus to determine the main prerequisites for practically useful permanent magnets. The computational HTS procedure is illustrated by results for hard-magnetic RE–TM phases like RETM5, RE2TM17 and RE2TM14B.
Wider implications. With our computational methodology we identified a new interesting RE–TM phase which was also confirmed experimentally. This indicates the great potential of computational modeling for finding a way out of the limited number of RE-based magnetic materials by extending the combinatorial possibilities from the known magnetic phases to the much wider conceivable configuration space of topologically close-packed phases.
1. Introduction
In recent years of rapid developments in the fields of electro-mobility and renewable-energy technologies, permanent magnetic materials composed of rare-earth (RE) and transition-metal (TM) elements have become of major importance due to their wide range of applications as hard-magnetic RE–TM phases for permanent magnets in electrical motors and generators, and various other functional components. However, the discovery of Nd2Fe14B, which currently shares first place with Ba/Sr ferrites as the most used material for permanent magnets, dates back to 1984 (Croat et al 1984, Herbst et al 1984). Due to the limited resources of RE elements and a latent supply monopoly on the international market, the goal is to discover and develop new hard-magnetic materials with lower RE content (ultimately RE-free magnets) and improved hard-magnetic performance (Kramers et al 2012). It is the purpose of this work to propose a fast and efficient computational strategy for the search for such novel hard-magnetic materials.
In recent reviews, e.g. by Kramers et al (2012), Coey (2011, 2012) or Gutfleisch et al (2011), different prospects and perspectives for the development of hard magnetic materials are discussed and the current state in the experimental and theoretical characterization of such materials is described. The large quantity of permanent magnets needed in contemporary technology, especially with the increased interest in alternative energy sources, transmission and conversions, also causes ecological concerns and economic problems, which are discussed in details by Alonso et al (2012).
The systematic search for novel hard magnetic materials is hardly achievable only via experimental discovery due to the very large combinatorial space spanned by the crystal structures and chemical compositions for compounds of RE, TM and interstitial (IS) elements. This difficulty is also connected with eventually high and unstable costs of raw materials on the world market and the time-consuming procedures of synthesis and characterization. In a very recent paper by Curtarolo et al (2013) a way toward an automated and efficient computational materials-design procedure was reviewed, and among the different areas of applications discussed magnetic materials were considered. This high-throughput screening (HTS) procedure combines a first-principles method of density functional theory (DFT) for computing the magnetic and thermodynamic properties of materials with the development of a large database of material parameters and the application of data mining procedures (Curtarolo et al 2013).
It is of great importance for the theoretical search for hard-magnetic materials to gain detailed insight into the most relevant physical mechanisms which determine the intrinsic properties of the hard-magnetic materials (Fähnle et al 1993). These intrinsic properties are spontaneous magnetization Ms, Curie temperature Tc and magnetocrystalline anisotropy energy (MAE), the latter characterized by, e.g., the constants K1 and K2 for uniaxial anisotropy. These properties are determined by local magnetic moments, exchange and spin–orbit interactions of electronic states on the atomic level. From the theoretical point of view, one can compute the local and total magnetic moments, exchange interactions as well as anisotropy constants which can deliver reasonable estimates for the desired intrinsic properties of the magnetic materials.
In almost all modern hard-magnetic materials the intrinsic material parameters are achieved as desired (Buschow 1991, Coey 1996) by combining TM and RE elements in particular proportions. This results in a large class of possible chemical compositions and crystal structures. In these compounds, the RE partner with 4f electron states is responsible for the high magnetic anisotropy, whereas the TM partner with 3d electron bands is responsible for the large magnetic moment and the ferromagnetic coupling which ensures a high Curie temperature. It is therefore well established that combining these two types of elements one can get materials with both high magnetic anisotropy and high Curie temperature (Fähnle et al 1993).
There are several prerequisites for the candidates for novel intermetallic RE–TM compounds which have to be fulfilled in order to apply these materials as practical permanent magnets with high energy density (Coey 1996). The use of permanent magnets in high temperature applications requires the candidate materials to have a Curie temperature well above the Tc = 590 K of Nd2Fe14B. The next important criterion concerns the magnetic anisotropy of intermetallic phases. To improve the anisotropy contribution of the crystal structure, preference is given to non-cubic, uniaxial-symmetry crystal structures such as tetragonal, rhombohedral or hexagonal ones. A positive anisotropy constant similar to the K1 = + 4.9 of Nd2Fe14B (Coey 1996) is desired, so that the c-axis is the easy direction. Furthermore, a magnetic saturation polarization Js = μ0Ms greater than 1.0 T should be achieved.
The purpose of this paper is to present a HTS procedure which assesses the computed local and total magnetic moments, exchange interactions as well as anisotropy constants in both a reasonably accurate and efficient manner to meet the above mentioned criteria for hard magnetic materials. The paper is organized as follows. In section 2 the computational procedure used for the investigation of permanent magnetic materials is described. In section 3 we present and discuss illustrative results for some common hard magnetic phases, namely RETM5, RE2TM14B and RE2TM17. The validation of the magnetic properties computed with the computationally fast but potential-shape-restricted tight-binding-linear muffin-tin-orbital-atomic-sphere-approximation (TB-LMTO-ASA) method by a potential-shape-unrestricted DFT method of high accuracy, namely the Vienna ab initio simulation package (VASP) (Kresse and Furthmüller 1996a, 1996b, Kresse and Joubert 1999), as well as computations of magnetic anisotropy, Curie temperature and phase stability of RE–TM compounds are presented in sections 4–7. A summary with some prospects for improvements of the proposed HTS methodology for the search for high-performance permanent magnetic materials concludes the paper.
2. Computational methods
The computational modeling and design of RE–TM intermetallic phases face three major problems associated with the nature of the constituting RE and TM elements and the choice of proper theoretical method. The first two problems were already described by Fähnle et al (1993) and are connected to: (i) the interplay between the localized magnetism of the 4f electrons of the RE atoms and the itinerant magnetism originating from 3d electrons of the TM atoms and (ii) the large unit cells, which have to be treated (in the case of Nd2Fe14B the unit cell consists of four formula units or 68 atoms). The third problem is the computational time needed to perform the HTS procedure for the search of possible intermetallic phases due to the large combinatorial space spanned by the variety of distributing RE, TM and IS elements in crystal structures.
2.1. High-throughput density-functional-theory calculations of the magnetic properties
Since there is a plethora of potential crystal structures and chemical compositions, there is a need for a fast, efficient and automated HTS procedure to find permanent magnetic materials. As already mentioned in the introduction, a comprehensive description of HTS procedures was provided by Curtarolo et al (2013). The practical implementation of HTS methods is a highly non-trivial task. Many variations are possible which depend on the choice of the first-principles methods software packages used as well as the data mining approaches. In recent years, there are only a few papers so far addressing the application of the HTS procedures for the search of permanent magnetic materials. For instance, Gilleßen and Dronkowski (2009, 2010) performed an extended combinatorial study with DFT on a set of 810 normal and inverse full Heusler phases. The authors investigated the thermodynamic stability and the ferromagnetism of a large class of possible Heusler alloys and identified a number of so far unknown stable magnetic phases as candidates for high density memory storage devices. Related to this theoretical HTS work, the group of Felser et al studied the Heusler phases by experimental HTS methods, succeeded in synthesizing several promising hard-magnetic phases experimentally and thus confirmed some theoretical predictions (Winterlik et al 2012, Gasi et al 2013, Kiss et al 2013).
The crucial step in the modeling of novel intermetallic RE–TM compounds as practical permanent magnets is to substitute the critical RE elements by other elements. This can be achieved by a systematic investigation of the magnetic properties of a large class of crystal structures and compositions. To tackle this problem, we have developed an automated HTS procedure to estimate the magnetic properties of RE–TM compounds. The work-flow diagram is shown in figure 1.
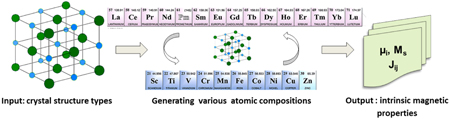
Figure 1. Work-flow diagram representing the automated HTS procedure for the search of new hard magnetic compounds.
Download figure:
Standard image High-resolution imageThe HTS procedure starts with a given crystal structure selected from a database of known crystal structure types, which belong to, e.g., the family of topologically close-packed phases (Seiser et al 2011). For the selected crystal structure, various chemical compositions of the possible RE, TM and IS elements are generated and their intrinsic magnetic properties such as total and local magnetic moments, and exchange integrals are computed with the computationally fast TB-LMTO-ASA method. For most promising candidate phases, the anisotropy constants (K1) and Curie temperatures (Tc) are then calculated in a subsequent step with the computationally accurate VASP code. All these computational tasks can be performed in an automated way more efficiently if one includes various restart options, the possibility for input-file correction as well as parallelization of TB-LMTO-ASA runs. The results of these HTS computations are compiled in a large materials database of intrinsic magnetic properties for intermetallic phases.
The crystal phases selected in this work (RE2TM14B, RE2TM17 and RETM5) to test the proposed HTS methodology cover a wide range of tetragonal, rhombohedral and hexagonal structure types. There exist further crystal structure types with RE:TM ratios like 1:12 and 3:29 which all can be derived from the basic hexagonal CaCu5 structure (see figure 2) as described by Burzo et al (1990) and Rao et al (1999).

Figure 2. Schematic representation of the structural interrelationships of RE–TM phases with RE:TM ratios 1:5, 1:12, 2:17 and 3:29.
Download figure:
Standard image High-resolution imageOur initial choice of elements was guided by the crustal abundance of the TM and RE elements and by the local magnetic moments of their elemental ferromagnetic phases. The most abundant and cheapest TM element is iron; it also possesses the largest local magnetic moment of 2.217 μB as present in bcc Fe (Skomski 2008). The most abundant RE elements are actually the 'light' ones with less than half-filled 4f electron shells, like cerium or neodymium. The magnetic moments of the light RE elements are oriented parallel to those of the TM (Blundell 2001). Therefore, for combinations of light RE elements and Fe, one expects larger magnetic moments of the constituting intermetallic phases. Although the 'heavy' RE elements have spins anti-parallel to those of the TM atoms and typically lead to lower total magnetic moments, their addition can increase the Curie temperature (Blundell 2001).
2.2. Fast computation of magnetic properties with the tight-binding-linear muffin-tin-orbital-atomic-sphere-approximation method
For the fast computation of the intrinsic magnetic properties of the magnetic materials, we use the LMTO method (Andersen 1975) in the ASA with the TB representation of LMTOs (Andersen and Jepsen 1984). All calculations were performed within the framework of the local-spin density approximation (LSDA) using a minimal spd basis set of LMTOs and the exchange-correlation functional of von Barth and Hedin (1972) in the parameterization of Moruzzi et al (1978). The ASA is a potential-shape approximation, for which the crystal is subdivided into space-filling and therefore slightly overlapping atomic spheres (the total unit-cell volume is equal to the sum of atomic-sphere volumes). Inside each sphere the effective crystal potential has spherical symmetry. This method yields accurate results for many physical quantities as long as the overlap of the atomic spheres is small, as in close-packed metals. In the choice of atomic-sphere radii for the different sets of RE, TM and IS elements, we rely on previous experience (Beuerle and Fähnle 1992) that the ratio of r(RE):r(TM):r(IS) = 1.35:1.00:0.70 yields results for magnetic properties which are very close to experimental ones.
Since the LSDA is valid for weakly correlated systems and the RE–TM systems have more strongly correlated, localized f-electrons in addition to delocalized d-electrons, the f-electrons are treated in the so called 'open-core' approximation (Brooks et al 1991a, 1991b), by imposing two constraints for the 4f charge and spin density entering the effective potential in LSDA. Within this approximation, one explicitly does not allow direct hybridization of f-electron states with other valence-electron states but we simulate the effect of 4f states below the valence band by imposing a non-zero logarithmic derivative to the radial 4f wave-functions at the atomic-sphere surfaces of the RE atoms. The constraint for the charge density fixes the number of electrons in the 4f core to the one of a free RE3+ ion. The constraint for the spin density fixes the magnetic spin moment of the 4f core to the value obtained from the atomic Russell–Saunders coupling for the projection of the free-ion 4f spin moment along the direction of the total 4f moment. In this way the strong Hund's rule correlations in the 4f shells are approximately accounted for. The magnetic moments calculated with the Russell–Saunders coupling often agree with experiment, although crystal-field splittings may also alter the moments. Although the interaction between the magnetism of the valence-band states and that of the 4f states occurs through the local exchange-correlation potential, there will effectively be a coupling between the RE 4f and TM 3d states in RE–TM compounds via hybridization between the RE 5d and TM 3d states (Brooks et al 1991a, 1991b). The corresponding numbers of spin-up and spin-down f-electrons used in the TB-LMTO-ASA calculations according to the frozen-core approximation can be found in Liebs (1992) and Blundell (2001).
Besides the total and local magnetic moments, we are also interested in the exchange parameters as quantities for comparison with experimental results and phenomenological models (Coey 1996), and particularly as model parameters for Monte Carlo simulations to determine critical temperatures (see section 6). To compute the exchange couplings in the RE–TM intermetallics we use the so called two-sublattice model (Fähnle et al 1993). Within this model, one divides the crystal lattice into two sublattices, one composed of RE elements and the other of TM elements only (see figure 2). If the crystal structure of interest has only one crystallographic site for RE atoms, there are two spin states with energies E1 and E2 (see figure 3) which need to be computed for every stoichiometry to get an estimate for the JRT integrals according to

where E1 and E2 are the total energies of the parallel and antiparallel spin states (see figure 3), SR is the magnetic spin moment of the RE sublattice, ST is the averaged magnetic spin moment of the TM sublattice, ZRT is the number of nearest neighboring TM atoms around the RE atom and ZR is the number of RE elements in the unit cell.
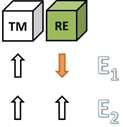
Figure 3. The two possible spins states of the two-sublattice model for a ferromagnetic RE–TM phase with only one crystallographic site for the RE element.
Download figure:
Standard image High-resolution imageThe determination of JRT is important for the following reasons (Fähnle et al 1993): (i) JRT is responsible for the transfer of the strong RE magneto-crystalline anisotropy to the TM sublattice due to the crystal-field coupling; (ii) at high temperatures JRT contributes to the transfer of the TM magneto-crystalline anisotropy (which then is larger than the RE anisotropy) to the RE sublattice; and (iii) JRT determines the thermal stability of the 4f moments, hence the very strong temperature dependence of the RE anisotropy and also the critical temperature Tc (Buschow 1991).
3. Results and discussion
In the following, results are presented separately for the RETM5, RE2TM14B and RE2TM17 structures which cover the best known RE:TM ratios of 1:5, 1:7 and 2:17.
3.1. Magnetic properties of RETM5 phases
The simplest intermetallic phase RETM5 considered here is chosen to illustrate the effect of substitutions on the magnetic properties. The compounds with the stoichiometry RETM5 crystallize in the hexagonal CaCu5-type structure shown in figure 4. For the calculation of the magnetic properties of this series we have applied experimental data for the atomic positions and lattice parameters of SmCo5 (Steinbeck et al 2001). In this structure, the Sm atom occupies the crystallographic 1a site and Co atoms enter the 2c and 3g sites (here and in the following, the crystallographic sites are denoted as Wyckoff positions of the space groups (for details see the International Tables for Crystallography at http://it.iucr.org/ or the Bilbao Crystallography Server at http://www.cryst.ehu.es/). The SmCo5 lattice parameters are kept constant (i.e. fixed length a and ratio c/a) during the HTS procedure for the entire series RETM5.

Figure 4. Illustration of the hexagonal CaCu5 structure of SmCo5 as an example of the RETM5 phases; crystallographic sites Sm(1a) are shown in blue, Co(2c) in red and Co(3g) in green; the thin black lines indicate the unit cell.
Download figure:
Standard image High-resolution imageOn the TM sites in REFe5 and RECo5 compounds the effect of mutual substitutions of Co and Fe atoms on the two sublattices is investigated. The calculated total magnetic moments are shown in figure 5. The RETM5 compounds with the light RE elements (Ce–Sm) possess higher magnetic moments than the ones with the heavy RE elements (Gd–Yb). (Note that Eu is a special case because it prefers a divalent ionic configuration over a trivalent one.) In the investigated series RECo5, RECo3Fe2, RECo2Fe3 and REFe5, maximal values for magnetic moments are found for RE = Pr, Nd.
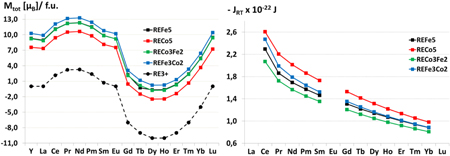
Figure 5. Total magnetic moments per formula unit (left) and exchange integrals (right) of the compounds from the REFe5−xCox series. The black circles and dashed line labeled RE3+ indicate the magnetic moments of free RE3+ ions.
Download figure:
Standard image High-resolution imageThe magnetic moments and exchange interactions in Sm(Fe,Co)5 have been previously investigated theoretically (Liu and Altounian 2011). In our work we extend the treatment to the entire series of 4f-elements. The partial substitutions in RECo5 with iron at crystallographic 2c or 3g positions have different effects on the magnetic properties in comparison to the pure RECo5 and REFe5 phases. Upon Co(2c) substitution with Fe, the total magnetic moment of RECo3Fe2 increases with values close to those of the only Fe-containing compounds REFe5. The partial substitution of Co(3g) with Fe, leading to REFe3Co2, results in a further improvement of the magnetic properties. The total magnetic moments of REFe3Co2 even exceed the magnetic moments of the REFe5 phases. This is because the local magnetic moment of Fe(3g) is highly reduced from about 2.4 μB in RECo3Fe2 to approximately 1.8 μB in REFe5. A similar tendency has been found by Liu and Altounian (2011), their LSDA + U approach yielded a reduction of the Fe-magnetic moment from 2.5 to 2.0 μB. From the results for the local magnetic moments in RECo5−xFex phases, we can conclude that simple linear interpolation assuming μ(Fe) = 2.217 μB and μ(Co) = 1.753 μB (Skomski 2008) to get an estimate for the magnitude of the magnetic moments of compounds is not generally applicable, since in some crystal structures some of these moments can be substantially quenched or enhanced. The better way to explore such tendencies is via systematic DFT computations.
The magnitude of the RE–TM exchange integrals JRT for the REFe5 and RECo5 compounds decreases monotonically with the atomic number of the RE elements (see figure 5). Larger JRT are found for RECo5 in comparison to REFe5. The Co(2c) substitutions with Fe lead to JRT values smaller than those of REFe5; the corresponding Co(3g) substitutions with Fe slightly increase the exchange-parameter values.
The dependence of the exchange integrals and magnetic moments on the unit cell size was investigated to check the applicability of the faster HTS procedure with lattice cell parameters kept fixed. For this purpose, the cell volume for each RETM5 compound was optimized by minimization of the total energy with respect to the unit-cell volume with the TB-LMTO-ASA method. The magnetic properties are compared in figure 6.

Figure 6. Change of the lattice parameters a (for fixed c/a ratio) with the atomic number of the RE element (left) and comparison of the magnetic properties for the optimized and fixed RETM5 unit-cell volumes (right). In the left graph, the dashed and dotted black lines are linear fits to LMTO and experimental values (Grazulis et al 2009, Villars and Cenzual 2012/13) of lattice parameters.
Download figure:
Standard image High-resolution imageThe optimized lattice parameter decreases almost linearly from Ce to Lu (as expected according to the well-known trend of RE elements called 'lanthanide contraction'). The total change in the lattice parameter, however, amounts to only about 0.132 Å for the whole series of 4f-elements. As one can see in the right panel in figure 6, the change in the cell volume affects the total magnetic moments and exchange integrals very weakly, which gives a justification for the fast HTS procedure employed by keeping lattice parameters constant along the RE series.
3.2. Magnetic properties of RE2TM17 compounds
The next challenge for the application of the developed HTS procedure is for intermetallic compound with several crystallographic sites for substitution, which extends substantially the possible variety of compositions. As an example we consider the RE2TM17 phases which exist in two different allotropic forms—the rhombohedral Th2Zn17-type structure (left panel of figure 7) and the hexagonal Th2Ni17-type one (right panel of figure 7) (Rama Rao et al 1999). The rhombohedral structure is stable for the light RE elements (from Ce to Gd), whereas for the heavy RE elements (from Gd to Lu) the hexagonal one is more stable. The compounds with RE = Gd, Tb and Y exist in both forms depending on the conditions for synthesis (Buschow 1977, Rama Rao et al 1999). It has also been found for the RE2TM17 compounds that some elements with small atomic radii (mainly hydrogen, carbon and nitrogen) can occupy IS sites of rhombohedral or hexagonal structures thus causing lattice expansion and improving the magnetic properties of the compound (Isnard et al 1990, 1994, Zhong et al 1990, Ibberson et al 1991, Beuerle and Fähnle 1992, Sun et al 1992). The 2:17-type samarium-cobalt magnets possess excellent intrinsic magnetic properties such as high saturation magnetization, high anisotropy fields and very high Curie temperature; the latter being even superior to Nd2Fe14B high-performance magnets (Schobinger et al 2002, Guo et al 2006). However, their high amount of the critical metal Co instead of Fe makes them very expensive.

Figure 7. Crystal structures of the rhombohedral Th2Zn17-type (left) and hexagonal Th2Ni17-type (right) allotropes of RE2Fe17 compounds.
Download figure:
Standard image High-resolution imageIn these compounds there are four crystallographic positions for the TM elements and either one position (for the rhombohedral structure) or two positions (for the hexagonal one) for the RE elements. In the ab initio screening procedure the atomic coordinates and lattice parameters for Nd2Fe17 (Isnard et al 1990) are used for the Th2Zn17-type and those of Gd2Fe17 (Knyazev et al 2006) for the Th2Ni17-type. Again as for the RETM5 phases, the lattice constant was kept fixed to the value of Nd2Fe17 for the entire series of RE elements and various TM substitutions when the magnetic properties were computed using the TB-LMTO-ASA method.
3.2.1. Magnetic properties of pure Fe- and Co-containing RE2TM17 phases
Figure 8 shows the total magnetic moments Mtot and the exchange integrals JRT for the pure Fe- and Co-containing phases of the entire series of RE elements. The results for the only Fe containing phases are shown in black, those for Co in red. One clearly sees that, like the RETM5 phases (cf figure 5), the RE2TM17 phases of Th2Zn17-type with light RE elements possess the larger total magnetic moments, with maxima at Pr and Nd. The Co-containing phases are characterized by lower magnetic moments, but their exchange integrals are larger. La, Eu and Lu are excluded from the computation of JRT since, according to the Russell–Saunders coupling, their RE3+ ions have magnetic moments of approximately zero.
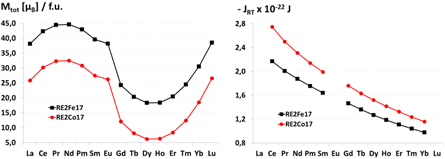
Figure 8. Computed total magnetic moments (left) and exchange integrals JRT (right) for the pure iron and cobalt containing RE2TM17 phases of Th2Zn17-type. The black lines represent the results for Fe, the red ones those for Co.
Download figure:
Standard image High-resolution imageAs can be seen from figure 8, the magnitude of the exchange integrals JRT for both RE2Fe17 and RE2Co17 decreases with increasing atomic number of the RE element. A similar tendency was already observed by Belorizky et al (1987) for various classes of RE–TM intermetallics and related to the variation of the 4f–5d exchange interactions. These interactions are larger for the light RE since the difference between the spatial extent of the 4f and 5d electrons is reduced.
Additional insight into magnetic properties of the RE2TM17 compounds can be obtained from the computed local magnetic moments. Figure 9 shows the changes in magnitude of the magnetic moments at the iron and cobalt sites of the TM-pure RE2TM17 phases.

Figure 9. Local magnetic moments of Fe in RE2Fe17 (left) and Co in RE2Co17 (right) at the four different crystallographic positions in the rhombohedral Th2Zn17-type structure for the whole series of RE elements.
Download figure:
Standard image High-resolution imageFrom figure 9 it is evident that the two TM elements Fe and Co interact quite differently with the RE sublattice, which leads to different ordering of the local magnetic moments at the four crystallographic 2c, 3d, 6f and 6h positions. For Fe the descending order of magnetic moments is μ(Fe-2c) > μ(Fe-6f) > μ(Fe-6h) > μ(Fe-3d) whereas for cobalt it is μ(Co-2c) > μ(Co-6f) > μ(Co-6h) > μ(Co-3d). This means that by exchanging Fe and Co at various crystallographic positions we can alter the magnetic properties in many ways.
3.2.2. Single and double substitutions of Fe by Co in Sm2Fe17
In the previous paragraph we described the magnetic properties of the purely Fe- and Co-containing RE2TM17 phases. But in the RE2TM17 phases, it is also possible to replace only some of the crystallographic TM positions with another TM element. To give an example, let us consider the Co and Mn substitution in Sm2Fe17, shown in figure 10. We chose this system as a representative case, since from all the RE2TM17 compounds Sm2Co17 is the most widely used hard magnetic phase. The results for Co substitutions are shown in blue, those for Mn in red. We can see that by adding small amounts of Co one just slightly lowers the total magnetic moments, thus approaching the moment of the purely Co-containing phase RE2Co17. Adding Mn leads to more pronounced lowering of the total magnetic moments; the total magnetic moments also approach that of the purely Mn-containing phase RE2Mn17. As concerns the exchange integrals JRT, usually the single Co substitutions lead to a slight increase and the single Mn substitutions to a large reduction of the JRT. The numerical values for the JRT integrals of the substituted Sm2Fe17−xCox compounds are in between those for Sm2Fe17 and Sm2Co17.
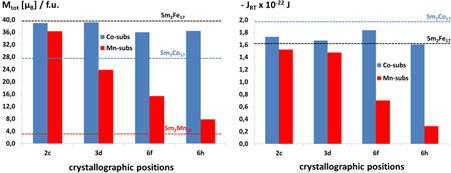
Figure 10. Computed values for the total magnetic moments Mtot (left) and exchange integrals JRT (right) for the ternary phases Sm2Fe17−xCox and Sm2Fe17−xMnx. The results for Co substitutions are shown in blue, those for Mn in red. The results for the binary systems are marked by dashed horizontal lines.
Download figure:
Standard image High-resolution imageIt is straightforward to extend this replacement procedure to two or more crystallographic sites and thus to consider the effects of double, triple, quadruple, etc substitutions and to analyze which ones more favorably lead to larger magnetic moments and exchange integrals. We have investigated, e.g., the effect of cobalt substitutions on two crystallographic positions, the results are summarized in figure 11 below.
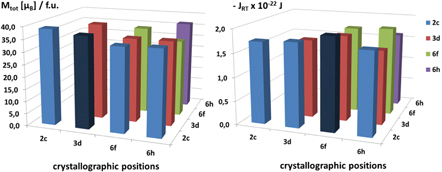
Figure 11. Two-dimensional charts representing the calculated total magnetic moments Mtot (left) and exchange integrals JRT (right) in Sm2Fe17−xCox for simultaneous substitutions of Fe by Co on two crystallographic positions. The diagonal elements correspond to single Co substitutions (cf figure 10) and are given here for comparison.
Download figure:
Standard image High-resolution imageSince Co has a lower magnetic moment per atom than Fe (μCo = 1.753 μB versus μFe = 2.217 μB for pure Co and Fe metals, respectively (Skomski 2008)), it is likely that the double Co substitution may lead to a further decrease of the total magnetic moments. However, a maximum of the total magnetic moments is achieved by double substitution at the pair of crystallographic 2c and 3d positions, which corresponds to the atomic composition Sm2Fe12Co5. As concerns the exchange parameters, a maximum value for JRT is obtained for the 2c–6f double substitutions, i.e. for Sm2Fe9Co8. This finding again supports the above statement that linear correlation of magnetic properties with chemical compositions is not generally valid and explicit DFT computation is better for analyzing and predicting magnetic properties.
3.3. Magnetic properties of Nd–Fe–B magnets and the RE2TM14B series
Since the discovery of the hard ferromagnetic compound Nd2Fe14B, for its development to the strongest available permanent magnet it has been a challenge to further improve its magnetic properties by compositional variations of the RE2Fe14B system. This may be achieved for instance by substituting Nd with another RE element, replacing totally or in part Fe by other TM elements like Co or Ni, or by exchanging B with other light IS (interstitial) atoms such as H, C and N. Because the combinatorial variety of possible intermetallic RE2TM14IS phases increases very rapidly, it is essential to obtain guidelines from theory for a systematic search for new materials.
The exact stoichiometry and crystal structure of Nd2Fe14B were determined independently and simultaneously by three research groups in 1984 (Givord et al 1984a, 1984b, Herbst et al 1984, Shoemaker et al 1984). Figure 12 displays the Nd2Fe14B unit cell. The crystal structure has tetragonal symmetry (space group P4z/mnm), and each unit cell contains four formula units, i.e. 68 atoms. There are six crystallographically distinct TM sites (16k1, 16k2, 8j1, 8j2, 4c and 4e), two different RE positions (4f and 4g) and one IS site (4g).
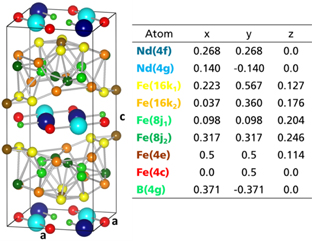
Figure 12. Unit cell of the Nd2Fe14B compound according to Herbst. The experimental lattice parameters are a = 0.88 nm, c = 1.22 nm. The c-axis was manually elongated to emphasize the puckering of iron layers as in Fähnle et al (1993).
Download figure:
Standard image High-resolution imageAnalogies with the hexagonal CaCu5 structure characterizing the permanent magnet SmCo5 and a variety of other RE–TM phases can be found in Herbst (1991) and references given therein.
In the ab initio screening of the magnetic properties in RE2TM14B compounds we used the experimental atomic positions and lattice parameters from Croat et al (1984). The results are presented in figure 13 and compared with those from previous theoretical (Liebs et al 1993) and experimental investigations (Herbst 1991).

Figure 13. Computed values for the total magnetic moments Mtot (left) and exchange integrals JRT (right) in RE2Fe14B; for comparison, experimental Mtot data in the left graph are from Herbst (1991), theoretical JRT data in the right graph from Liebs et al (1993).
Download figure:
Standard image High-resolution imageThe computed results from the HTS procedure for the total magnetic moments show very good agreement with the experimental values for both RE2Fe14B and RE2Co14B. The largest discrepancy for the total magnetic moments is found for Ce2Fe14B and Sm2Co14B, respectively. This difference can be attributed to the tendency of Ce and Sm to form mixed valence intermetallics, which could lead to a deviation from the RE3+ configurations used in the calculations. The calculated JRT integrals also follow the same tendency as those found in the paper of Fähnle et al (1993) and Liebs et al (1993). The good agreement with experiment indicates again that one can compute reliable estimates for the magnetic moments with the fast TB-LMTO-ASA method.
4. Accurate density functional theory calculations with no potential-shape approximation
The results from the fast ab initio screening procedure with the TB-LMTO-ASA method reveal most of the features in the magnetic properties of the investigated RE–TM intermetallic phases. However, the examples provided in this paper are for well-known RE–TM phases. If we want to go beyond this and predict new phases as candidates for permanent magnetic materials we have to check the accuracy of the TB-LMTO-ASA method with DFT methods beyond the potential-shape restricting atomic-sphere approximation and to be able to compute the Curie temperature (Tc) and the magnetocrystalline anisotropy constant (K1).
We have investigated some selected systems by performing spin-polarized calculations using VASP (versions 4 and 5) (Kresse 1993, Kresse and Hafner 1993, Kresse and Furthmüller 1996a,1996b). Starting from experimental values, with VASP the lattice parameters and atomic positions are fully relaxed, and then the magnetic properties are calculated. In the LSDA calculations projector augmented wave potentials provided in VASP were used. After checking for convergence of the total energy differences for several systems, an energy cutoff of 400 eV for the plane-wave basis was selected for all the calculations. The k-points distribution was checked for convergence as well (e.g. for Nd2Fe14B system with 68 atoms in the unit cell a 5 × 5 × 4 Monkhorst-pack grid was used). Convergence criteria for the structure relaxation were set to 10−5 eV and 0.01 eV Å−1 for the energies and forces, respectively.
As concerns the accuracy of the TB-LMTO-ASA method for predicting the magnetic moments and the exchange interactions of the RE–TM intermetallics, we have compared some results of this method with those obtained by the full-potential LSDA calculations with VASP. The results for the hexagonal Nd2Fe17 and Nd2Fe17N3 compounds are summarized in figure 14.
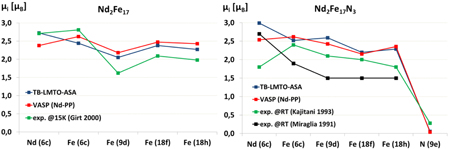
Figure 14. Comparison of the local magnetic moments for Nd2Fe17 (left) and Nd2Fe17N3 (right) calculated with TB-LMTO-ASA and VASP programs with reported experimental values (Miraglia et al 1991, Kajitani et al 1993, Girt et al 2000).
Download figure:
Standard image High-resolution imageThere is in general very good agreement between the results computed with both the fast DFT method and the accurate one, and with the experimental magnetic moments for both Nd2Fe17 and Nd2Fe17N3 phases, as can be seen from figure 14. The addition of nitrogen is expected to lead to a dilation of the lattice and thus to higher magnetic moments, which is well reproduced by both VASP and TB-LMTO-ASA.
5. Estimating the magnetocrystalline anisotropy energy with Vienna ab initio simulation package
The MAE is determined with VASP by doing a set of calculations starting with the self-consistent DFT density and Kohn–Sham wave functions obtained with the magnetization oriented in the z-direction (easy axis [001]) and gradually changing the direction of the spin quantization axis from this direction to the [100] or [110] direction, respectively, without recalculation of the wave functions. Total energies per unit cell are calculated for a series of orientations of the quantization axis, and then a polynomial function in the rotation angles with respect to the z-axis is fitted to the total-energy data. The form of the polynomial function depends on the symmetry of the crystal. For Nd2Fe14B the MAE is expressed by (Givord et al 1984a, 1984b)
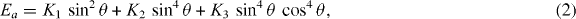
where K1, K2, K3 are the anisotropy constants, θ and φ are the angles of magnetization with respect to [001] and [100], respectively.
Note that for Nd2Fe14B at a transition temperature of 135 K a reorientation of the magnetic easy axis from a low-temperature orientation of θ = 32.3° inclined to [001] to an orientation parallel to the [001] direction takes place (Otani et al 1987, Wolfers et al 1996). This means that the experimental MAE curves at temperatures below 135 K have a minimum away from the [001] direction (figure 15, green curve at 4.2 K), and only for temperatures above this transition temperature does the easy axis coincide with the [001] direction (figure 15, red curve at 200 K). Thus, the theoretical MAE curves (and the derived K1 and K2 constants) calculated with the procedure implemented in VASP (figure 15, blue curve), where the easy axis is set in the [001] direction, are to be compared with the experimental data above the transition temperature.

Figure 15. The dependence of the MAE per unit cell on the orientation of the quantization axis with respect to the tetragonal c-axis for Nd2Fe14B. The blue line indicates the result of our VASP calculation. The green and red lines are from measurements under and above the reorientation transition temperature of the crystal, respectively. These data are reported in Otani et al (1987). Additional experimental (Givord et al 1984a, 1984b, Hirosawa et al 1986) and calculated data (Kitagawa and Asari 2010) for the MAE for a change of the magnetization direction from [001] to [100] are marked by symbols at θ = 90°.
Download figure:
Standard image High-resolution imageThe values for the anisotropy constants K1 and K2 obtained from the fits of equation (3) to the curves for small angles in the vicinity of the easy axis [001] are given in table 1 for different changes in the magnetization direction ([001]–[100] and [001]–[110]). They are compared with experimental data found in the literature for two temperature ranges, and with a value estimated from theoretical crystal-field parameters A20 calculated with a full-potential linear-muffin-tin-orbital method (FP-LMTO) (Hummler and Fähnle 1996).
Table 1. Magnetocrystalline anisotropy constants calculated with VASP for RE2Fe14B compounds with RE = Ce, Pr and Nd.
| Compound | Anisotropy constant (MJ m−3) | Calculated [001]–[100] | Calculated [001]–[110] | Experimental values | Calculated FP-LMTO-LSDA | |||
|---|---|---|---|---|---|---|---|---|
| 4.2 K | 270–300 K | |||||||
| Ce2Fe14B | K1 | 2.10 | 2.07 | 1.65a | – | 1.44a | – | |
| Pr2Fe14B | K1 | 4.85 | 4.89 | 2.35a | – | 5.5a | – | |
| Nd2Fe14B | K1 | 6.18 | 6.26 | −6.5b | −8.59c | 3.7b | 5.15c | 6.08d |
| B | K2 | 2.06 | 2.09 | 14b | 23.3c | 1.5b | 0.69c | – |
Corresponding experimental values are taken from: aSagawa (1987) at 300 K. bOtani et al (1987) at 4.2 K. cPastushenkov et al (2005) at 270 K. dThis theoretical value is estimated from theoretical crystal-field parameters A20 for the two Nd sites in Nd2Fe14B calculated by Hummler and Fähnle (1996).
For Nd2Fe14B the differences between our calculated values and the experimental values around room temperature are within the spread between the different experimental results. Our calculated values for K1 have less than 2% difference to the one obtained from the calculated A20 values from Hummler and Fähnle (1996). Similarly good agreement holds between our calculated values for K1 for Pr2Fe14B and Ce2Fe14B and the corresponding experimental ones reported at room temperature. Moreover, our calculated values for the total MAE obtained by changing the magnetization axis completely from [001] to [100] almost coincide with the values reported by Givord et al (1984a, 1984b) and Hirosawa et al (1986), and they are in better agreement with the experimental data than other calculated values using linear combination of pseudo-atomic-orbitals within the LSDA + U approximation (Kitagawa and Asari 2010).
We conclude that the VASP method allows to calculate values for anisotropy constants of hard-magnetic RE–TM phases that reproduce in general the typical behavior of complex magnetic materials, and that it has predictive power for the evaluation of the MAE by complete tilting (θ = 90°) of the magnetization direction.
6. Estimate of the Curie temperature (Tc)
Another major challenge in the computational search for new candidates for permanent magnetic materials is to get a reasonable estimate of the Curie temperature (Tc), which is essentially determined by the interatomic exchange interactions. The two-sublattice model considered above provides a good approach to determine the exchange interaction between RE and TM sublattices. However, for TC it is the exchange interaction between the TM atoms which is more relevant, and therefore the distance-dependent TM–TM exchange integrals between TM atoms on different sublattices have to be calculated. The problem of calculating the magnetic TM–TM exchange interactions and determining TC therewith for RE–TM phases was addressed recently by Lukoyanov et al for Gd2Fe17 in its rhombohedral and hexagonal lattice structures (Lukoyanov et al 2009). They used the calculated values of exchange parameters for the first- and second-neighbor coordination spheres in both structure types and estimated TC on the basis of Weiss' mean-field theory.
Alternatively to the mean-field approach, TC can be determined as well from the exchange parameters by means of numerical finite-temperature Monte Carlo simulations. In this work, we chose this procedure. The computation of the pairwise TM–TM exchange interactions, Jij, needed therefore was done with the method proposed by Liechtenstein et al (1987). This method provides a rigorous expression for the pair exchange parameters of the classical Heisenberg model

applied to crystals, and the parameters are obtained by LSDA calculations. The total magnetic state of all atoms is described by the set of normalized spin vectors {si} (the subscript i is the atom index). In order to determine the exchange parameters Jij one must calculate the total energy for small deviations of a single magnetic moment and a pair of magnetic moments from the ferromagnetic ground-state configuration. This results in energy Ei for a spin configuration si and energy Eij for a spin-pair configuration sij, where the superscript indicates the tilted spins. The exchange interaction Jij is calculated from
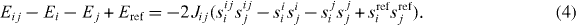
For small periodic unit cells one has to divide the resulting Jij by the number of occurrences of the interaction due to periodic images of spins. This approach, however, is valid for T = 0 K only. To consider finite temperatures with DFT one needed to know the temperature dependence of the LSDA exchange-correlation functional which is not known yet (Liechtenstein et al 1987).
In order to reduce the number of necessary DFT calculations of tilted spin states for a given set of exchange interactions, the symmetry of the crystal structure was exploited to determine a pairwise non-symmetric subset of interactions. Where necessary the space group of the crystal structure was detected (Hannemann et al 1988). The energies of the tilted spin states were calculated with VASP. Since the local magnetic moments are integrals of the magnetization in a site centered sphere, they cannot be set directly and the spin orientations were constrained with a penalty contribution to the total energy expression.
A serial Monte Carlo program for a general crystal structure was developed using the Metropolis et al (1953), Swendsen and Wang (1987) and Wolff (1989) methods for the spin models of Ising (1925) and Heisenberg (1928). In contrast to the Metropolis method the latter algorithms do not flip single spins but a cluster of spins, which were calculated with the Hoshen–Kopelman algorithm (Hoshen et al 1979). The Curie temperature was determined as the intersection of the fourth-order cumulant of magnetization–temperature curves for different sized systems (Shan-Ho and Salinas 1998).
The exchange interactions for bcc Fe have been calculated for interatomic distances up to 8.2 Å, i.e. the 12th nearest neighborhood, and resulted in a Curie temperature of 1100 K. This is in good agreement with the experimental value of 1043 K (Yousuf et al 1986). For SmCo5 with interactions up to only 3.2 Å the resulting calculated TC value of 2500 K does not yet reproduce well the experimental Curie temperature of 985 K (Tie-Song et al 1991). Therefore further neighborhoods of interactions need to be taken into account. This is still work in progress.
7. Thermodynamic stability of rare-earth–transition-metal phases
The theoretical characterization of new phases with favorable magnetic properties is only the first step for the real discovery of a new magnetic material. Before experimental work for the synthesis of a promising candidate begins, modeling should give some confidence as well about the expectable thermodynamic stability of the new material. In the following we give only a concise outlook on the current extension of our HTS procedure in this direction.
A first evaluation of the stability can be done by comparing the formation energies calculated by DFT methods like VASP. This procedure and its possibilities are illustrated by discussing the thermodynamic stability in the Ce–Fe system. Phases based on Ce and Fe show potential for alternative hard magnetic materials, since both these RE and TM elements are abundant and comparatively cheap. In addition, the magnetic properties of CexFey phases calculated with TB-LMTO-ASA (not shown here) are also in an acceptable range.
In the binary phase diagram of the Ce–Fe system (Okamoto 2008) there are only stable phases identified for four compositions: Ce, CeFe2, Ce2Fe17 and Fe. There are additional reports about the synthesis of CeFe5 in the literature (Jepson and Duwez 1955). We calculated the formation energies of the known binary phases with respect to the pure metallic elemental phases of fcc Ce and bcc Fe. The results for the formation energy for the different phases including CeFe12 are shown in figure 16. None of the phases CeFe5, CeFe12 and Ce2Fe17 seem to be stable in the sense that they are not part of the stability convex hull of the system (Hildebrandt and Glasser 1994). Phases with more than 66.6% content of Fe would decompose into CeFe2 and Fe. Particularly for Ce2Fe17 this result is a consequence of the neglect of the contribution of temperature-dependent terms to the total energy of the material. Actually, preliminary calculations including vibrational-energy contributions for the binary Ce–Fe system predict stability for Ce2Fe17 starting at a temperature of about 600 K, where this phase becomes a vertex of the convex hull (in the experimental phase diagram this change occurs at about 1200 K).

Figure 16. Calculated formation energies of the CeFex phases as a function of the iron content. Higher negative values imply more stable phases. The resulting convex hull is drawn with dashed lines.
Download figure:
Standard image High-resolution imageAs a strategy for the stabilization of CeFe12, which has the most favorable RE:TM proportion within the binary phases, some of the Fe atoms are substituted by other elements. The existence of known stable phases of related ternary phases RETM10Si2, like YFe10Si2 and GdFe10Si2 (Mooij and Buschow 1988), points to the possibility of trying the stabilization with a double substitution of Fe by Si. This substitution results in a formation energy of −0.37 eV per atom, far lower than the formation energy of CeFe2 in the binary Ce–Fe system, which makes the ternary phase apparently more stable than the binary one. Unfortunately this potentially new ternary phase CeFe10Si2 has also to compete with all other known binary and ternary phases of the ternary Ce–Fe–Si configuration space.
Finding in the literature all known mixed phases for a given combination of several elements is a cumbersome task. To make it easier, a screening tool was designed to screen the most common crystallographic data bases, namely Inorganic Crystal Structure Database (Bergerhoff and Brown 1987), Crystallography Open Database (Grazulis et al 2009) and Pearson's Crystal Data (Villars and Cenzual 2012/13): given the list of elements of interest, the software finds all binary, ternary and more-components mixed phases, automatically eliminates duplicate entries and lists the atomic compositions, crystal structures and the literature citations. In a future advanced version this HTS tool will also be capable of creating the input files for a chosen DFT code to run the calculation of the formation energies.
Figure 17 shows the result from our VASP calculations for the Ce–Fe–Si system, including the interesting new candidate CeFe10Si2. In the framework of the approximate DFT total energies, the proposed phase is part of the convex hull of the ternary system and in this way also predicted to be stable. Yet more accurate DFT calculations are needed to confirm this result since the new phase is located in the vicinity of experimentally reported phases of the composition Ce2Fe(15.0–16.5)Si(2–0.5) and CeFe(9.5–8)Si(3.5–5). These two substitutional phases are represented in our calculation by single compositions CeFe9Si4 and Ce2Fe15Si2, and have probably a high entropic contribution to their total energy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 17. Calculated phase diagram of the Ce–Fe–Si system based on formation energies calculated with VASP. The color scale gives the formation energies in eV per atom. Black lines connect the convex hull vertices.
Download figure:
Standard image High-resolution image{kind=link}
Further improvement is needed in this procedure to include in an automatized way more exact estimations for the total energies of the involved systems, taking into account further terms like vibrational free energies and configurational entropies for phases with substitutional elements.
8. Summary and outlook
In this paper we presented an efficient and robust HTS methodology for the systematic computation of the intrinsic magnetic properties of various intermetallic RE–TM compounds by using fast and reliable first-principle methods based on DFT. The HTS procedure was tested for a selection of known hard magnetic phases such as RETM5, RE2TM17 and RE2TM14B. The effects of different RE and TM substitutions on the total magnetic moments and exchange integrals were thoroughly investigated with the fast TB-LMTO-ASA method for each intermetallic phase. It was shown that for the selected as promising hard magnetic materials it is possible to compute the Curie temperature (Tc) and magnetocrystalline anisotropy constants (K1) with more accurate DFT calculations using VASP and thus to determine the main prerequisites for practically useful permanent magnetic materials. The task of predicting structural stability of the RE–TM phases was also addressed using VASP. Our described HTS approach is able to determine the main guiding principles in the selection of proper crystal structures and atomic compositions, which can lead to new candidates for hard magnetic phases. In a next step, we are going to apply the proposed HTS procedure to predict the relative stability of the RE–TM phases and screen more general classes of topologically close-packed phases for promising candidates of permanent magnetic materials.
Acknowledgments
This work was supported by the Cooperative Project 'Suche nach neuen hartmagnetischen Phasen mit hoher Energiedichte (REleaMag)' (grant number 03X4626A) funded by the German Federal Ministry of Education and Research (BMBF). We are grateful to Professor Olivier Isnard for critical reading of the manuscript. Some of the authors (AM, LK, AG, TS and TE) would like to express their sincere thanks to the crew of Center for Computational Materials Science of the Institute for Materials Research, Tohoku University for their continuous support of the SR16000 supercomputing facilities. We especially want to thank Professor Yoshiyuki Kawazoe (Tohoku University, Japan) for many stimulating and highly valuable discussions.