


Abstract
We employ positron annihilation lifetime spectroscopy (PALS) to measure the temperature dependent fluctuations in a local free volume of water confined in a variety of mesoporous materials with different pore sizes and morphologies. We demonstrate that this unconventional approach can be used to probe the dynamics and glass transition temperature, Tg, of water in confinement. In the simplest case of water confined in a 13X molecular sieve (d = 7.4 Å), the confinement is so severe that it precludes crystallisation altogether, and we measure a Tg = 190 ± 2 K. We also show that temperature dependent PALS measurements can be used to probe the glass transition of water molecules confined in matrices with larger pore diameters (SBA-15, KIT-6 and Vycor glass), where the confinement is expected to be less severe.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Corrections were made to this article on 23 October 2014. The name of the author F Kleitz was corrected.
1. Introduction
The glass transition temperature, Tg, of water is one of the long-standing problems in condensed matter physics, motivating a large plethora of studies to date [1–6]. Recently, this topic was elegantly discussed by Capaccioli and Ngai [6], who presented a multi-faceted approach towards solving this problem. The difficulty in determining Tg for water lies in its high propensity for crystallisation. At ambient pressure, water exists in the crystalline state in the temperature range of 150–235 K (where 235 K is the homogenous nucleation temperature of water [5]), creating an experimentally inaccessible region for studying the dynamics of its amorphous phase [3, 5, 6]. A common way of suppressing crystallisation is by confining water in nano-meter-sized cavities in materials such as porous glasses, clays and zeolites [5, 7]. It has been shown that with decreasing pore diameter, confined water crystallizes at increasingly lower temperatures [7] until a critical diameter is reached below which the freezing of water is completely suppressed [8–10]. Studies have also indicated that two types of water molecules exist in confinement: interfacial ones, in contact with the pore wall and internal ones, located in the pore center, only interacting with other water molecules [7, 10–12]. The thickness of the interfacial layer (4–6 Å) [7] is insensitive to the pore diameter, therefore, a larger fraction of pore water is situated in this layer with decreasing pore size [7]. Water confined at the nano-scale is known to have different structural, dynamic and thermodynamic properties from that of bulk water due to strong surface interactions, truncation of the bulk correlation length and a modified hydrogen bond network [13]. Nano-confinement has been shown to frustrate the hydrogen bonding of water molecules, allowing a different quantum ground state of the electron–proton system compared to the bulk [14, 15].
Notwithstanding, confinement of water at the nanometer length scale offers the possibility of exploring its properties at temperatures well below the bulk homogenous nucleation temperature [7]. Understanding the glass transition dynamics of water in confined geometries also has a significant relevance for a number of technological (e.g. catalytic and separation processes) and biological processes [16]. For example, in living organisms water molecules often exist in highly confined geometries (e.g. protein hydration water and intracellular water) [16–18], and Tg is often recognised as the boundary above which biological systems function [19–21]. For these reasons the dynamics of water in confinement have been studied by a large number of experimental techniques, including nuclear magnetic resonance [5, 22], dielectric spectroscopy [18, 23–29] and neutron scattering [27, 30–35]. However, the interpretation of such relaxation measurements has not been straightforward, since for supercooled water in confinement the most pronounced mode of relaxation is the more local, non-cooperative  -relaxation [18, 26, 36]. Due to the reduction in length scale and the modification of the hydrogen bond network in confinement [13], the cooperative α-relaxation (associated with the glass transition) for water appears to be either vanishingly small [26] or non-existent [23, 24, 31, 32], making the assignment of Tg non-trivial. Furthermore, supercooled water in confinement typically shows extremely weak and broad changes in heat capacity [26], making the unequivocal assignment of Tg very cumbersome. Perhaps some of the more definitive studies have been those performed using adiabatic calorimetry [9–11]. For water confined in silica gel and porous MCM-41, the presence of two distinct glass transition temperatures (from interfacial and internal water, respectively), has been confirmed by observing systematic spontaneous enthalpy relaxation effects [9–11]. It was noted that the interfacial water molecules do not crystallize and exhibit a Tg = 114–125 K (depending on the pore size), whilst the internal water molecules remain in the supercooled state in pores with diameters up to 20 Å with Tg = 160–165 K (depending on the pore size) [9–11].
-relaxation [18, 26, 36]. Due to the reduction in length scale and the modification of the hydrogen bond network in confinement [13], the cooperative α-relaxation (associated with the glass transition) for water appears to be either vanishingly small [26] or non-existent [23, 24, 31, 32], making the assignment of Tg non-trivial. Furthermore, supercooled water in confinement typically shows extremely weak and broad changes in heat capacity [26], making the unequivocal assignment of Tg very cumbersome. Perhaps some of the more definitive studies have been those performed using adiabatic calorimetry [9–11]. For water confined in silica gel and porous MCM-41, the presence of two distinct glass transition temperatures (from interfacial and internal water, respectively), has been confirmed by observing systematic spontaneous enthalpy relaxation effects [9–11]. It was noted that the interfacial water molecules do not crystallize and exhibit a Tg = 114–125 K (depending on the pore size), whilst the internal water molecules remain in the supercooled state in pores with diameters up to 20 Å with Tg = 160–165 K (depending on the pore size) [9–11].
In this paper, we re-visit the problem of Tg of supercooled water in confinement through a less conventional approach, utilising the unique probe of positron annihilation lifetime spectroscopy (PALS). PALS is a well established technique extensively used for the study of local free volume and dynamics in a wide range of glass-forming systems, from simple liquids to synthetic polymers [37–41]. Over the past decade, PALS has been successfully used to study a variety of simple molecular glass formers both, in confinement [42] and in bulk [43–49]. Such studies have shown that model-free analysis of the temperature dependence of the ortho-positronium (o-Ps) lifetime,  (reflecting the local free volume) reveals the existence of several characteristic temperatures [44, 47, 48]. One of these characteristic temperatures can be identified as the glass transition temperature,
(reflecting the local free volume) reveals the existence of several characteristic temperatures [44, 47, 48]. One of these characteristic temperatures can be identified as the glass transition temperature,  [48, 49], while the two other have been labelled as
[48, 49], while the two other have been labelled as 
 1.2–1.3
1.2–1.3  and
and 
 1.4–1.7
1.4–1.7  [44, 48, 49]. These PALS temperatures can be empirically related to various parameters associated with the cooperative α-relaxation [44, 50]. For example, at
[44, 48, 49]. These PALS temperatures can be empirically related to various parameters associated with the cooperative α-relaxation [44, 50]. For example, at  the o-Ps lifetime is equal to the mean α-relaxation time, while at
the o-Ps lifetime is equal to the mean α-relaxation time, while at  the characteristic α-relaxation time is
the characteristic α-relaxation time is  10−6 s [44]. Furthermore, the thermal variations of
10−6 s [44]. Furthermore, the thermal variations of  can be correlated with the changes in the dielectric α-relaxation time [44, 47] and its non-Arrhenius/'fragile' characteristics [50]. For example, a greater rate of thermal increase in
can be correlated with the changes in the dielectric α-relaxation time [44, 47] and its non-Arrhenius/'fragile' characteristics [50]. For example, a greater rate of thermal increase in  above Tg has been shown to correspond to higher kinetic 'fragility' for a number of glass-forming liquids [50, 51]. Finally, it has been shown that the temperature dependent changes in the free volume fraction probed by PALS also show striking similarities with the temperature dependence of the mean square atomic displacements,
above Tg has been shown to correspond to higher kinetic 'fragility' for a number of glass-forming liquids [50, 51]. Finally, it has been shown that the temperature dependent changes in the free volume fraction probed by PALS also show striking similarities with the temperature dependence of the mean square atomic displacements,  , evidenced by incoherent neutron scattering [52, 53]. All of this hints to the existence of a close relationship between the local free volume probed by PALS and the dynamics of simple molecular glass formers.
, evidenced by incoherent neutron scattering [52, 53]. All of this hints to the existence of a close relationship between the local free volume probed by PALS and the dynamics of simple molecular glass formers.
Here, we use temperature dependent PALS measurements to probe the glass-transition behaviour of supercooled water confined in four different types of porous materials: 13X zeolite molecular sieve (d = 7.4 Å), pseudo-one dimensional SBA-15 highly ordered silica (d = 70.3 Å), three-dimensional cubic KIT-6 highly ordered silica (d = 70.3 Å) and porous Vycor glass (d = 70.0 Å). We demonstrate that in the simplest case, for the 13X molecular sieve, where the confinement is so severe that crystallisation of water is believed to be completely suppressed, our positron lifetime measurements can be utilised to unambiguously probe its glass transition dynamics, yielding a Tg = 190 ± 2 K. More interestingly, we also observe the same glass transition features for water confined in larger pores, where the confinement is expected to be significantly less severe.
2. Materials and methods
2.1. Preparation of the confining materials
All experiments were carried out using ultra-pure Milli-Q water (Millipore, Bedford MA, USA) with a resistivity of 18.2 MΩ cm measured at 298 K. Several materials, with different structures and average pore sizes were used for the confinement experiments. The 13X zeolite molecular sieve (d = 7.4 Å) was purchased from Sigma-Aldrich (Sigma-Aldrich, Gillingham, UK). Details of the network structure of the 13X zeolite can be found in [54]. Highly ordered mesoporous silica materials with exceptionally narrow pore size distributions [55, 56], designated as SBA-15 (d = 70.3 Å, pseudo-one-dimensional system) and KIT-6 (d = 70.3 Å, two interwoven networks of branched cylindrical channels), were synthesized via cooperative self-assembly of an amphiphilic structure-directing agent (poly(alkylene oxide) triblock copolymer, Pluronic P123) and an inorganic precursor (tetraethylorthosilicate), under acidic conditions. Full details of the synthesis of these materials can be found elsewhere [57]. The pore size analysis of these highly ordered mesoporous glasses was performed by high-resolution nitrogen (T = 77.4 K) adsorption/desorption isotherm measurements, performed with an Autosorb-1-MP adsorption instrument (Quantachrome Instruments, Florida, USA) in a relative pressure, ( ), range from 1 × 10−6 to 1, see [57]. The characterisation data of the two mesoporous silica materials are summarized in table 1. The SBA-15, KIT-6 and 13X zeolite materials used for the PALS measurements were degassed at 400 K in a vacuum oven for 48 hours, after which they were re-hydrated in Milli-Q water. Vycor glass with an average pore diameter, d = 70.0 Å (Dow Corning, Midland, Michigan, USA) was cut into
), range from 1 × 10−6 to 1, see [57]. The characterisation data of the two mesoporous silica materials are summarized in table 1. The SBA-15, KIT-6 and 13X zeolite materials used for the PALS measurements were degassed at 400 K in a vacuum oven for 48 hours, after which they were re-hydrated in Milli-Q water. Vycor glass with an average pore diameter, d = 70.0 Å (Dow Corning, Midland, Michigan, USA) was cut into  2 mm thick disks for the PALS experiments. The disks were cleaned prior to the experiments by boiling in 30
2 mm thick disks for the PALS experiments. The disks were cleaned prior to the experiments by boiling in 30 hydrogen peroxide, rinsing thoroughly in Milli-Q water, followed by drying in a vacuum oven at 420 K for 48 h (see table 1). The samples were then re-hydrated in Milli-Q water. Excess surface water was removed prior to using all samples. The water contents of the samples (wt
hydrogen peroxide, rinsing thoroughly in Milli-Q water, followed by drying in a vacuum oven at 420 K for 48 h (see table 1). The samples were then re-hydrated in Milli-Q water. Excess surface water was removed prior to using all samples. The water contents of the samples (wt ), expressed as
), expressed as  (where mw—mass of adsorbed water and mm—mass of the dry matrix) were determined in triplicate by oven drying at 473 K for 48 h. The degree of pore filling, ϕ, of the SBA-15 and KIT-6 materials was calculated from
(where mw—mass of adsorbed water and mm—mass of the dry matrix) were determined in triplicate by oven drying at 473 K for 48 h. The degree of pore filling, ϕ, of the SBA-15 and KIT-6 materials was calculated from  , where vp is the specific pore volume determined by nitrogen adsorption at 77.4 K and ρw = 0.997 g cm−3 is the bulk liquid density of water at 298 K.
, where vp is the specific pore volume determined by nitrogen adsorption at 77.4 K and ρw = 0.997 g cm−3 is the bulk liquid density of water at 298 K.
Table 1. Characterization parameters for the mesoporous materials used: aaverage pore diameter, bspecific pore volume and cwater content.
| Confining matrix | d ( Å)a | vp (cm3 g−1) b | Qw (wt%)c |
|---|---|---|---|
| SBA-15 | 70.3 | 0.79 | 21 |
| KIT-6 | 70.3 | 0.91 | 22 |
| Vycor | 70.0 | n.d. | 20 |
| 13X zeolite | 7.4 | n.d. | 23 |
2.2. Positron annihilation lifetime spectroscopy
PALS experiments proceed by injecting a positron into the material being tested and measuring the length of time until that positron annihilates with one of the materialʼs electrons, producing γ-rays. In molecular materials, a significant fraction of the injected positrons form a meta-stable positron–electron bound state, known as positronium (Ps), which requires regions of local free volume (low electron density) to form and exist. Ps has two spin states:  -positronium (p-Ps), a singlet state with zero spin angular momentum and
-positronium (p-Ps), a singlet state with zero spin angular momentum and  -positronium (o-Ps), a triplet state with unit spin angular momentum. The lifetime of the more abundant and longer lived
-positronium (o-Ps), a triplet state with unit spin angular momentum. The lifetime of the more abundant and longer lived  -positronium, o-Ps, is environment dependent and it can be related to the size of the free volume hole using a simple quantum mechanical model. In this model, o-Ps is assumed to be localized in a spherical potential well of an infinite depth and a radius,
-positronium, o-Ps, is environment dependent and it can be related to the size of the free volume hole using a simple quantum mechanical model. In this model, o-Ps is assumed to be localized in a spherical potential well of an infinite depth and a radius,  [58, 59]
[58, 59]
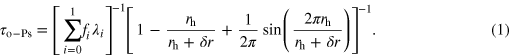
Here,  is the free volume hole radius and the positronium has an overlap with molecules within a layer of
is the free volume hole radius and the positronium has an overlap with molecules within a layer of  of the potential wall [58, 59].
of the potential wall [58, 59].  is the fraction of positronium with spin i (1/4 for
is the fraction of positronium with spin i (1/4 for  -Ps, spin 0; 3/4 for
-Ps, spin 0; 3/4 for  -Ps, spin 1) and λi is the corresponding annihilation rate in vacuum (0.125 ns−1 and 142 ns−1, respectively) [37, 38]. In order to avoid overemphasis of this spherical hole model, here we present our data in terms of
-Ps, spin 1) and λi is the corresponding annihilation rate in vacuum (0.125 ns−1 and 142 ns−1, respectively) [37, 38]. In order to avoid overemphasis of this spherical hole model, here we present our data in terms of  rather than rh. The positron annihilation lifetime experiments were performed using a conventional fast–fast coincidence system (fully described elsewhere [38, 42]) whose resolution can be modeled as a Gaussian function (175 ps, full-width at half maximum) convoluted with two exponential wings. 22Na was used a source of positrons and it was prepared by evaporating carrier-free 22NaCl solution between 7.5 μm thick sheets of Kapton foil. Measurements were taken over the temperature range of 100–340 K, and spectra at each temperature were collected over a minimum period of 2 h to generate at least 2.5 million events per spectrum. The source correction was 14.8% of the total spectrum, with two components measured, 0.41 ns (I = 90.2%) and 3.62 ns (I = 9.8%). These components were accounted for prior to the analysis of the spectra. The measured source correction (14.8% of the total spectrum) was slightly higher than a standard source correction, typically 7–14% (depending on the thickness of Kapton used) [60, 61]. This is due to the fact that the source used in our experiments was reinforced with an extra layer of Kapton as a precaution, since it was used for water. The lifetime spectra were analysed assuming a three discrete component fit [37, 38] using the fitting routine Life Time (version 9.1) [62], which performs a weighted non-linear least squares fit of the following model function to the experimental spectra
rather than rh. The positron annihilation lifetime experiments were performed using a conventional fast–fast coincidence system (fully described elsewhere [38, 42]) whose resolution can be modeled as a Gaussian function (175 ps, full-width at half maximum) convoluted with two exponential wings. 22Na was used a source of positrons and it was prepared by evaporating carrier-free 22NaCl solution between 7.5 μm thick sheets of Kapton foil. Measurements were taken over the temperature range of 100–340 K, and spectra at each temperature were collected over a minimum period of 2 h to generate at least 2.5 million events per spectrum. The source correction was 14.8% of the total spectrum, with two components measured, 0.41 ns (I = 90.2%) and 3.62 ns (I = 9.8%). These components were accounted for prior to the analysis of the spectra. The measured source correction (14.8% of the total spectrum) was slightly higher than a standard source correction, typically 7–14% (depending on the thickness of Kapton used) [60, 61]. This is due to the fact that the source used in our experiments was reinforced with an extra layer of Kapton as a precaution, since it was used for water. The lifetime spectra were analysed assuming a three discrete component fit [37, 38] using the fitting routine Life Time (version 9.1) [62], which performs a weighted non-linear least squares fit of the following model function to the experimental spectra
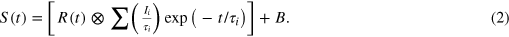
Here, i = 1, 2, 3 are the components attributed to the annihilation of p-Ps, free positron and o-Ps, respectively, weighted by the relevant intensity Ii, (where  = 1). R(t) is the resolution function, where t is time and B is the background [62].
= 1). R(t) is the resolution function, where t is time and B is the background [62].

Figure 1. Temperature dependence of the o-Ps lifetime,  , for glycerol in confinement and in bulk, measured during a cooling run. The solid lines represent linear regression fits to the data (see table 2) and all correlation coefficients, R2
, for glycerol in confinement and in bulk, measured during a cooling run. The solid lines represent linear regression fits to the data (see table 2) and all correlation coefficients, R2  0.98. The point of intersection of the low and high temperature branches reflects the change in dynamics at the glass transition, and it is denoted as
0.98. The point of intersection of the low and high temperature branches reflects the change in dynamics at the glass transition, and it is denoted as  . For a detailed discussion on the possible origins of the changes in the thermal response of
. For a detailed discussion on the possible origins of the changes in the thermal response of  at
at  and
and  , see [44, 47–49]. Inset: Comparison of the Tg values determined by PALS and DSC measurements for a number of simple molecular glass formers: propylene carbonate (PC), o-terphenyl (OTP), bis[m-(m-phenoxy)phenoxyphenyl] ether (6PO), 1,1-bis(paramethoxy-metamethylphenyl)-cyclohexane (BMMPC) and glycerol (GLY). Data are collated from previous studies [42–44].
, see [44, 47–49]. Inset: Comparison of the Tg values determined by PALS and DSC measurements for a number of simple molecular glass formers: propylene carbonate (PC), o-terphenyl (OTP), bis[m-(m-phenoxy)phenoxyphenyl] ether (6PO), 1,1-bis(paramethoxy-metamethylphenyl)-cyclohexane (BMMPC) and glycerol (GLY). Data are collated from previous studies [42–44].
Download figure:
Standard image High-resolution image3. Results and discussion
We begin our discussion by briefly illustrating how the dynamics of simple molecular glass formers are reflected in the temperature dependent positron lifetime measurements. In figure 1 we present temperature dependent PALS data for glycerol. Glycerol is chosen here as an example because of its good glass-forming ability and due to the fact that, like water, it is a strongly hydrogen bonded liquid. Measurements are shown both for bulk and in confinement (Vycor glass, d = 70.0 Å), over a wide temperature range across the glass transition. In both cases, our data are in excellent agreement with previously reported PALS measurements for glycerol [42, 47]. The observed temperature dependence of the o-Ps lifetime measured for bulk reflects the typical behaviour of molecular glass formers as they pass through their glass transition [43–49]. Three characteristic temperatures can be identified in the thermal response of  measured in bulk glycerol, which according to previously described notation can be identified as
measured in bulk glycerol, which according to previously described notation can be identified as  ,
,  and
and  [44, 48, 49]. Here, we focus on the changes in o-Ps lifetime associated with the glass transition, but the possible origins of the changes in the thermal response of
[44, 48, 49]. Here, we focus on the changes in o-Ps lifetime associated with the glass transition, but the possible origins of the changes in the thermal response of  at
at 
 240 K and
240 K and 
 290 K are discussed in detail by Barto
290 K are discussed in detail by Barto et al [44, 47–49]. At temperatures below
et al [44, 47–49]. At temperatures below  , the molecules are 'frozen' into the glassy state and
, the molecules are 'frozen' into the glassy state and  reflects the average size of the static free volume elements. The slight increase in
reflects the average size of the static free volume elements. The slight increase in  with increasing temperature mirrors the thermal expansion of the local free volume in the glassy matrix due to the anharmonicity of the molecular vibrations. As the temperature increases and glycerol passes through its glass transition, the molecular motions increase rapidly resulting in a strong
with increasing temperature mirrors the thermal expansion of the local free volume in the glassy matrix due to the anharmonicity of the molecular vibrations. As the temperature increases and glycerol passes through its glass transition, the molecular motions increase rapidly resulting in a strong  dependence, indicating a rapid increase in the local free volume. The temperature dependence of
dependence, indicating a rapid increase in the local free volume. The temperature dependence of  measured for glycerol at high and low temperatures can be fitted well by linear regression (see table 2). The point of intersection between the low and the high temperature branches reflects the change in dynamics at the glass transition, providing a measure of the glass transition temperature (
measured for glycerol at high and low temperatures can be fitted well by linear regression (see table 2). The point of intersection between the low and the high temperature branches reflects the change in dynamics at the glass transition, providing a measure of the glass transition temperature ( = 185 ± 2 K), as confirmed by calorimetric measurements [42–44]. There is essentially a direct correspondence between the values of the glass transition temperature determined by PALS,
= 185 ± 2 K), as confirmed by calorimetric measurements [42–44]. There is essentially a direct correspondence between the values of the glass transition temperature determined by PALS,  and those measured by differential scanning calorimetry (DSC),
and those measured by differential scanning calorimetry (DSC),  , as shown in the inset of figure 1 for a range of simple molecular glass formers [42–44]. Following this line of argument, the glass transition temperature of glycerol confined in Vycor was found to be
, as shown in the inset of figure 1 for a range of simple molecular glass formers [42–44]. Following this line of argument, the glass transition temperature of glycerol confined in Vycor was found to be  = 190 ± 2 K, in agreement with previous measurements [42]. The difference in the glass transition temperature measured in confinement compared to bulk reflects the difference in glycerol dynamics due to the interplay between finite size effects and the complex surface interactions between the glycerol molecules and the pore walls of the confining Vycor matrix [42]. Finally, it is interesting to note that the PALS data for bulk glycerol presented in figure 1 closely mirror the thermal response of the mean square atomic displacements,
= 190 ± 2 K, in agreement with previous measurements [42]. The difference in the glass transition temperature measured in confinement compared to bulk reflects the difference in glycerol dynamics due to the interplay between finite size effects and the complex surface interactions between the glycerol molecules and the pore walls of the confining Vycor matrix [42]. Finally, it is interesting to note that the PALS data for bulk glycerol presented in figure 1 closely mirror the thermal response of the mean square atomic displacements,  , probed by incoherent neutron scattering on similar timescales4
(
, probed by incoherent neutron scattering on similar timescales4
( 1 ns using the IN10-IN16 spectrometer) [63]. In fact, the two characteristic PALS temperatures,
1 ns using the IN10-IN16 spectrometer) [63]. In fact, the two characteristic PALS temperatures,  = 185 K and
= 185 K and 
 240 K closely match the temperatures at which the thermal response of
240 K closely match the temperatures at which the thermal response of  has been previously shown to change [63]. Similar observations were also reported for a poly(methyl methacrylate) glass, where the changes in
has been previously shown to change [63]. Similar observations were also reported for a poly(methyl methacrylate) glass, where the changes in  were found to be strongly correlated with the changes in free volume fraction [52, 53]. This further highlights the close relationship between free volume and the dynamics of glass-forming systems.
were found to be strongly correlated with the changes in free volume fraction [52, 53]. This further highlights the close relationship between free volume and the dynamics of glass-forming systems.
Table 2.
 Gradients of the linear regression lines,
Gradients of the linear regression lines,  , fitted to the temperature dependent lifetime data for glycerol in bulk and in confinement. Typical standard error: 2 × 10−4. bThe temperature at which the two linear branches intersect, denoted as
, fitted to the temperature dependent lifetime data for glycerol in bulk and in confinement. Typical standard error: 2 × 10−4. bThe temperature at which the two linear branches intersect, denoted as  . Typical standard error: 2. dGradient of the Tg—scaled temperature dependence of
. Typical standard error: 2. dGradient of the Tg—scaled temperature dependence of  for
for  ,
,  =
=  [50]. Typical standard error 0.1.
[50]. Typical standard error 0.1.
a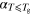 (ns K−1) (ns K−1) |
b (K) (K) |
c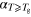 (ns K−1) (ns K−1) |
d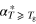 (ns) (ns) |
|
|---|---|---|---|---|
| Bulk | 1.0 × 10−3 | 185 | 8.1 × 10−2 | 1.5 |
| Vycor | 1.1 × 10−3 | 190 | 9.0 × 10−3 | 1.7 |
In figure 2(a) we show the  temperature dependence measured during heating and cooling scans for water confined in the 13X zeolite molecular sieve (d
temperature dependence measured during heating and cooling scans for water confined in the 13X zeolite molecular sieve (d  7.4 Å) and in bulk. In both cases, no significant differences in
7.4 Å) and in bulk. In both cases, no significant differences in  were observed between the heating and cooling scans. For this reason, the majority of the discussion in this paper shall be based on the changes in
were observed between the heating and cooling scans. For this reason, the majority of the discussion in this paper shall be based on the changes in  measured during the cooling scans. Over the entire temperature range, the o-Ps lifetimes measured for water confined in the 13X molecular sieve are longer than those measured in bulk water, with the largest differences in
measured during the cooling scans. Over the entire temperature range, the o-Ps lifetimes measured for water confined in the 13X molecular sieve are longer than those measured in bulk water, with the largest differences in  reported for T
reported for T  273 K. Similar observations have been previously reported for simple molecular glass formers in confinement [42] (e.g. for glycerol, as already shown in this paper), reflecting the altered dynamics due to the restricted geometry and strong surface interactions. The temperature dependence of
273 K. Similar observations have been previously reported for simple molecular glass formers in confinement [42] (e.g. for glycerol, as already shown in this paper), reflecting the altered dynamics due to the restricted geometry and strong surface interactions. The temperature dependence of  observed for water confined in the 13X molecular sieve solely reflects the changes in dynamics of the confined phase, since we do not observe an additional o-Ps lifetime component from annihilation within the bulk of the confining matrix. If the changes in the o-Ps lifetime did contain an additional component from annihilation in the molecular sieve, it would either be resolved or, if it is experimentally un-resolvable, the χ2 of the fit would improve as the o-Ps lifetime measured approaches that of the confining matrix, neither of which was observed (as shown in figure 2(b)). Since the focus of this paper is on the low temperature behaviour of water, we shall not discuss the temperature dependence of
observed for water confined in the 13X molecular sieve solely reflects the changes in dynamics of the confined phase, since we do not observe an additional o-Ps lifetime component from annihilation within the bulk of the confining matrix. If the changes in the o-Ps lifetime did contain an additional component from annihilation in the molecular sieve, it would either be resolved or, if it is experimentally un-resolvable, the χ2 of the fit would improve as the o-Ps lifetime measured approaches that of the confining matrix, neither of which was observed (as shown in figure 2(b)). Since the focus of this paper is on the low temperature behaviour of water, we shall not discuss the temperature dependence of  measured at temperatures higher than 273 K (the melting temperature of bulk water, Tm) in much detail. It is, however, worth mentioning that at these temperatures water is in the liquid state, and the behaviour of positronium is phenomenologically different as it manifests the ability of self-trapping [37, 38, 64]. At temperatures above Tm, the o-Ps lifetime measured for water decreases as a function of temperature, in agreement with previous observations, and the origin of this phenomenon has been discussed by several authors [65–68]. One proposed explanation relates the observed decrease in the o-Ps lifetime to the chemical reactions (e.g.
measured at temperatures higher than 273 K (the melting temperature of bulk water, Tm) in much detail. It is, however, worth mentioning that at these temperatures water is in the liquid state, and the behaviour of positronium is phenomenologically different as it manifests the ability of self-trapping [37, 38, 64]. At temperatures above Tm, the o-Ps lifetime measured for water decreases as a function of temperature, in agreement with previous observations, and the origin of this phenomenon has been discussed by several authors [65–68]. One proposed explanation relates the observed decrease in the o-Ps lifetime to the chemical reactions (e.g.  -to-
-to- conversion and Ps oxidation) of Ps with its intra-track radiolytic products (e.g. OH radicals and H3O+) [65, 68]. These chemical reactions at the nanosecond timescale are diffusion controlled processes, whose respective rate constants increase with temperature [65, 68], thus explaining the observed decrease in
conversion and Ps oxidation) of Ps with its intra-track radiolytic products (e.g. OH radicals and H3O+) [65, 68]. These chemical reactions at the nanosecond timescale are diffusion controlled processes, whose respective rate constants increase with temperature [65, 68], thus explaining the observed decrease in  .
.

Figure 2. (a) Temperature dependence of the o-Ps lifetime measured for water confined in a 13X molecular sieve and in bulk, during cooling and heating scans. The solid lines represent linear regression fits to the data (see table 3) and all correlation coefficients, R2  0.98.
0.98.  of supercooled water confined in the 13X molecular sieve is identified as the point of intersection of the two branches in the thermal response of
of supercooled water confined in the 13X molecular sieve is identified as the point of intersection of the two branches in the thermal response of  . The melting temperature of bulk water, Tm, is also shown for reference. (b) χ2 parameter of the non-linear least squares fit of the experimental lifetime spectra measured for water confined in the 13X molecular sieve at various temperatures.
. The melting temperature of bulk water, Tm, is also shown for reference. (b) χ2 parameter of the non-linear least squares fit of the experimental lifetime spectra measured for water confined in the 13X molecular sieve at various temperatures.
Download figure:
Standard image High-resolution imageFigure 2(a) shows that for  273 K, there is a striking difference in the
273 K, there is a striking difference in the  temperature dependence measured for bulk and confined water. For bulk water, we observe a discontinuous decrease in
temperature dependence measured for bulk and confined water. For bulk water, we observe a discontinuous decrease in  at Tm = 273 K, signifying the formation of crystalline ice. Given the slow cooling rates during the PALS experiments, the bulk water sample is undoubtedly completely crystalline at temperatures below 273 K. Our lifetime measurements for bulk water are in excellent agreement with those previously reported by Eldrup et al [69], showing no significant differences between
at Tm = 273 K, signifying the formation of crystalline ice. Given the slow cooling rates during the PALS experiments, the bulk water sample is undoubtedly completely crystalline at temperatures below 273 K. Our lifetime measurements for bulk water are in excellent agreement with those previously reported by Eldrup et al [69], showing no significant differences between  measured for mono- and poly-crystalline ice, confirming the high reproducibility of the data. The o-Ps lifetime of 0.6 ns measured for bulk water for temperatures up to about 180 K is compatible with o-Ps annihilating from a pseudo-delocalized state within spaces with dimensions equivalent to the inter-planar channels which exist in crystalline ice [16, 69]. The o-Ps lifetime remains constant over this temperature range, reflecting the negligible thermal expansion of the ice lattice. At temperatures above
measured for mono- and poly-crystalline ice, confirming the high reproducibility of the data. The o-Ps lifetime of 0.6 ns measured for bulk water for temperatures up to about 180 K is compatible with o-Ps annihilating from a pseudo-delocalized state within spaces with dimensions equivalent to the inter-planar channels which exist in crystalline ice [16, 69]. The o-Ps lifetime remains constant over this temperature range, reflecting the negligible thermal expansion of the ice lattice. At temperatures above  K,
K,  becomes temperature dependent, whilst remaining significantly shorter than the o-Ps lifetimes measured for water in confinement. This temperature dependence of
becomes temperature dependent, whilst remaining significantly shorter than the o-Ps lifetimes measured for water in confinement. This temperature dependence of  has been previously reported and may be ascribed to the creation of di- and tri-vacancies in thermal equilibrium [69, 70]. Trapping of Ps in these vacancies becomes apparent at a threshold temperature, related to the enthalpy of vacancy formation [69, 70]. The size of these cavities confining Ps does not change significantly as a function of increasing temperature, resulting in the observed weak temperature dependence of
has been previously reported and may be ascribed to the creation of di- and tri-vacancies in thermal equilibrium [69, 70]. Trapping of Ps in these vacancies becomes apparent at a threshold temperature, related to the enthalpy of vacancy formation [69, 70]. The size of these cavities confining Ps does not change significantly as a function of increasing temperature, resulting in the observed weak temperature dependence of  for temperatures up to Tm.
for temperatures up to Tm.
For water confined in the 13X molecular sieve, we do not observe an abrupt decrease in  signifying a shifted crystallisation temperature in the vicinity of 273 K. Instead, the temperature dependence of
signifying a shifted crystallisation temperature in the vicinity of 273 K. Instead, the temperature dependence of  reflects the features seen for a typical molecular glass-former as it approaches and passes through its glass transition. The water molecules in this matrix are present in a severely confined state, where possibly all water is interfacial and thus completely amorphous, as confirmed by a number of previous studies [9–11]. For example, Oguni et al, demonstrated that crystallisation of water is completely suppressed in pores of diameters smaller than 20 Å [9–11]. The observed temperature dependence of
reflects the features seen for a typical molecular glass-former as it approaches and passes through its glass transition. The water molecules in this matrix are present in a severely confined state, where possibly all water is interfacial and thus completely amorphous, as confirmed by a number of previous studies [9–11]. For example, Oguni et al, demonstrated that crystallisation of water is completely suppressed in pores of diameters smaller than 20 Å [9–11]. The observed temperature dependence of  below 273 K thus reflects the dynamics of water in its supercooled state. Following the line of thought outlined in figure 1 for glycerol [47], the glass transition temperature,
below 273 K thus reflects the dynamics of water in its supercooled state. Following the line of thought outlined in figure 1 for glycerol [47], the glass transition temperature,  , of supercooled water molecules confined in the 13X zeolite can be identified as the point of intersection of the low and high temperature branches of the thermal response of
, of supercooled water molecules confined in the 13X zeolite can be identified as the point of intersection of the low and high temperature branches of the thermal response of  (see table 3). Thus, we find that
(see table 3). Thus, we find that  = 190 ± 2 K. It is interesting to note that
= 190 ± 2 K. It is interesting to note that  lies in close proximity to the crossover temperature reflecting the deviation from Arrhenius temperature dependence of the dielectrically observed
lies in close proximity to the crossover temperature reflecting the deviation from Arrhenius temperature dependence of the dielectrically observed  -like process previously observed for water confined in the same 13X molecular sieve [25]. By comparing neutron scattering [33] and dielectric spectroscopy [25] measurements, Swenson et al suggested that this could be attributed to the merging of the
-like process previously observed for water confined in the same 13X molecular sieve [25]. By comparing neutron scattering [33] and dielectric spectroscopy [25] measurements, Swenson et al suggested that this could be attributed to the merging of the  -like process with an extremely weak [26] or even invisible α-relaxation [33].
-like process with an extremely weak [26] or even invisible α-relaxation [33].
Table 3.
 Gradients of the linear regression lines,
Gradients of the linear regression lines,  , fitted to the temperature dependent lifetime data for water confined in various matrices. Typical standard error: 2 × 10−4. bThe temperature at which the two linear branches intersect, denoted as
, fitted to the temperature dependent lifetime data for water confined in various matrices. Typical standard error: 2 × 10−4. bThe temperature at which the two linear branches intersect, denoted as  . dGradient of the Tg-scaled temperature dependence of
. dGradient of the Tg-scaled temperature dependence of  for
for  ,
,  =
=  [50]. Typical standard error 0.1.
[50]. Typical standard error 0.1.
| Confining matrix | a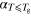 (ns K−1) (ns K−1) |
b (K) (K) |
c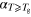 (ns K−1) (ns K−1) |
d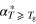 (ns) (ns) |
|---|---|---|---|---|
| 13X Zeolite | 1.5 × 10−3 | 190 ± 2 | 1.1 × 10−2 | 2.1 |
| SBA-15 | 1.2 × 10−3 | 189 ± 3 | 9.8 × 10−3 | 2.0 |
| KIT-6 | 2.3 × 10−3 | 191 ± 1 | 1.2 × 10−2 | 2.1 |
| Vycor | 1.8 × 10−3 | 190 ± 2 | 1.2 × 10−2 | 2.2 |
Having successfully demonstrated that temperature dependent PALS measurements can be utilised to unambiguously probe Tg of water in severe confinement, next we extend our study to water confined in matrices with larger pore diameters, where both internal and interfacial water molecules are likely to be present [9–11]. In figure 3(a) we show lifetime measurements for water confined in a range of mesoporous matrices with different morphologies and pore diameters of the order of 70 Å. Once again, we note that the changes in o-Ps lifetime solely reflect the changes in the dynamics of the confined water molecules since in all cases we do not resolve an extra lifetime component from annihilation with the confining matrices, nor do we observe any improvements in the χ2 of the fit, as illustrated in figure 3(b). It is evident from figure 3(a) that the  temperature dependences for water confined in all the matrices with larger pore diameters are all very similar, and in all cases no abrupt discontinuity in
temperature dependences for water confined in all the matrices with larger pore diameters are all very similar, and in all cases no abrupt discontinuity in  is observed to signify a shifted crystallisation in the neighbourhood of 273 K. This is interesting, since previous studies suggest that for water confined in pores of these sizes, while the interfacial water molecules remain in a supercooled state and exhibit a glass transition, the internal water molecules tend to crystallize at temperatures below 260 K [7, 9–11].
is observed to signify a shifted crystallisation in the neighbourhood of 273 K. This is interesting, since previous studies suggest that for water confined in pores of these sizes, while the interfacial water molecules remain in a supercooled state and exhibit a glass transition, the internal water molecules tend to crystallize at temperatures below 260 K [7, 9–11].

{kind=link}
{kind=link}
Figure 3. (a) Temperature dependent PALS measurements for water confined in a variety of mesoporous materials. The solid lines represent linear regression fits to the data (see table 3) and all correlation coefficients, R2  0.98.
0.98.  of supercooled water confined in the matrices is identified as the point of intersection of the two branches in the thermal response of
of supercooled water confined in the matrices is identified as the point of intersection of the two branches in the thermal response of  . (b) The χ2 parameter of the non-linear least squares fit of the experimental lifetime spectra.
. (b) The χ2 parameter of the non-linear least squares fit of the experimental lifetime spectra.
Download figure:
Standard image High-resolution image{kind=link}
The lack of a signature associated with the crystallisation of water confined in these matrices could be attributed to the fact that the pores are only partially filled due to the preparation method. For example, the water contents in the SBA-15 and KIT-6 silica matrices correspond to degrees of pore filling of 0.27 and 0.24, respectively. DSC studies have shown that whilst freezing and melting of water in completely filled pores gives rise to a single peak, complex freezing behaviour can be observed in partially filled pores [7, 71]. Up to three exothermic peaks, attributed to the different states of liquid in the pore space can be resolved in the freezing scans, while the melting scans still only show a single endothermic peak [7, 71]. Such studies of water confined in SBA-15 silica matrices (d = 77 Å) have shown that for similar degrees of pore filling, the intensity of all (exothermic and endothermic) peaks is extremely weak [71], which could explain the lack of a clear signature of crystallization/melting in the PALS data. Furthermore, the lack of any abrupt decreases in  measured for water confined in these matrices, may hint to a possible preference of Ps for the amorphous interfacial layer or the gradual freezing of any internal water, or indeed a combination of both.
measured for water confined in these matrices, may hint to a possible preference of Ps for the amorphous interfacial layer or the gradual freezing of any internal water, or indeed a combination of both.
It is clear from figure 3 that as for water confined in the 13X molecular sieve, the o-Ps lifetimes measured at temperatures below 273 K are consistent with annihilation from a localized state existing in the free volume elements of the supercooled liquid; rather than a pseudo-delocalized state within the interplanar channels in crystalline ice [69]. Furthermore, the temperature dependencies of  defined by the gradients,
defined by the gradients,  , of the low and the high temperature branches are indeed very similar to those measured for water confined in the 13X zeolite, where no crystalline fraction of water is expected to be present (see table 3). Following from this, the temperatures at which the two linear branches intersect can be identified as the glass transition temperatures,
, of the low and the high temperature branches are indeed very similar to those measured for water confined in the 13X zeolite, where no crystalline fraction of water is expected to be present (see table 3). Following from this, the temperatures at which the two linear branches intersect can be identified as the glass transition temperatures,  of water confined in the various matrices (see table 3). Given the fact that the pores of these matrices are underfilled, and that generally Tg of molecular glass formers (including water) [9–11, 42] often only changes by a few degrees as a function of pore diameter, it is not surprising to see that we do not observe significant differences in
of water confined in the various matrices (see table 3). Given the fact that the pores of these matrices are underfilled, and that generally Tg of molecular glass formers (including water) [9–11, 42] often only changes by a few degrees as a function of pore diameter, it is not surprising to see that we do not observe significant differences in  . As an aside, it is worth briefly discussing the gradients of the temperature dependence of
. As an aside, it is worth briefly discussing the gradients of the temperature dependence of  above
above  measured for water in confinement. It was previously shown that steeper gradients of the Tg-scaled temperature dependence of
measured for water in confinement. It was previously shown that steeper gradients of the Tg-scaled temperature dependence of  above Tg,
above Tg,  , can be related to a higher kinetic 'fragility' for a number of bulk glass-forming liquids [50, 51]. This suggests that the local free volume measured by PALS mirrors the thermal variations of the α-relaxation [50, 51]. For example, 'fragile' glass-forming liquids such as o-terpheyl show
, can be related to a higher kinetic 'fragility' for a number of bulk glass-forming liquids [50, 51]. This suggests that the local free volume measured by PALS mirrors the thermal variations of the α-relaxation [50, 51]. For example, 'fragile' glass-forming liquids such as o-terpheyl show 
 4.7 (estimated from [50]), while 'stronger' glasses such as propylene glycol and glycerol show
4.7 (estimated from [50]), while 'stronger' glasses such as propylene glycol and glycerol show 
 1.6 and 1.5, respectively [50]. Assuming the same argument holds for glass-forming liquids in confinement, the
1.6 and 1.5, respectively [50]. Assuming the same argument holds for glass-forming liquids in confinement, the  values presented in table 3 can be related to the 'fragility' of the confined water phase. It is clear from table 3 that
values presented in table 3 can be related to the 'fragility' of the confined water phase. It is clear from table 3 that 
 2.0 for water in confinement is very similar to that of glycerol, both in confinement:
2.0 for water in confinement is very similar to that of glycerol, both in confinement: 
 1.7 and in bulk:
1.7 and in bulk: 
 1.5 (see table 2), as well as that of bulk propylene glycol:
1.5 (see table 2), as well as that of bulk propylene glycol: 
 1.6 [50]. Water confined in the various matrices studied, therefore, appears to show a thermal behaviour of
1.6 [50]. Water confined in the various matrices studied, therefore, appears to show a thermal behaviour of  typical of 'stronger' glass-forming liquids (rather than 'fragile' glasses such as o-terpheyl), in agreement with the recent conclusions of Capaccioli and Ngai for bulk water [6].
typical of 'stronger' glass-forming liquids (rather than 'fragile' glasses such as o-terpheyl), in agreement with the recent conclusions of Capaccioli and Ngai for bulk water [6].
Finally, it is interesting to note that the glass transition temperature of water in confinement around 190 K found by PALS is consistent with the recent NMR study of water confined in MCM-41 [22], indicating a cross-over  190 K reflecting the change in the temperature dependence of the β-relaxation process in response to the glass transition of interfacial water.
190 K reflecting the change in the temperature dependence of the β-relaxation process in response to the glass transition of interfacial water.  also closely agrees with the findings from dielectric spectroscopy and DSC on hydration water in proteins [17] , where the geometrical confinement or binding of water molecules to the protein surface prevent crystallisation. For example, it has been shown that the relaxation time of the dielectrically observed
also closely agrees with the findings from dielectric spectroscopy and DSC on hydration water in proteins [17] , where the geometrical confinement or binding of water molecules to the protein surface prevent crystallisation. For example, it has been shown that the relaxation time of the dielectrically observed  -like process of hydration water in a purple membrane protein shows a crossover reflecting the deviation from Arrhenius temperature dependence at
-like process of hydration water in a purple membrane protein shows a crossover reflecting the deviation from Arrhenius temperature dependence at  K [17, 18]. This combined with an endothermic process observed by DSC [18], indicated that Tg is within the range of 190–200 K [17]. These observations further highlight the close relationship between the local free volume probed by PALS and the dynamics probed by techniques such as dielectric spectroscopy and NMR. One could envisage that the thermal density fluctuations relaxed through the more local, non-cooperative
K [17, 18]. This combined with an endothermic process observed by DSC [18], indicated that Tg is within the range of 190–200 K [17]. These observations further highlight the close relationship between the local free volume probed by PALS and the dynamics probed by techniques such as dielectric spectroscopy and NMR. One could envisage that the thermal density fluctuations relaxed through the more local, non-cooperative  -relaxatinon for supercooled water in confinement lead to changes in the local free volume distribution which can be accurately reflected in our temperature dependent PALS measurements. Thus, PALS is an invaluable direct probe for the glass transition dynamics of water in confinement which neatly complements the indirect observations from other dynamic techniques.
-relaxatinon for supercooled water in confinement lead to changes in the local free volume distribution which can be accurately reflected in our temperature dependent PALS measurements. Thus, PALS is an invaluable direct probe for the glass transition dynamics of water in confinement which neatly complements the indirect observations from other dynamic techniques.
4. Final remarks
To summarise, in this paper we show that the glass transition dynamics of water confined in a variety of mesoporous materials can be probed via the temperature dependent fluctuations in local free volume measured by PALS. We demonstrate that this unconventional approach can be used to unambiguously determine Tg of water in confined geometries. In the 13X molecular sieve (d = 7.4 Å), crystallisation is expected to be completely suppressed and we measure a glass transition temperature of the supercooled water at  = 190 ± 2 K. Finally, we show that temperature dependent PALS measurements can also be used to probe Tg of water even when the confinement is significantly less severe, i.e. when confined in the pores of Vycor glass, SBA-15 and KIT-6 silicas.
= 190 ± 2 K. Finally, we show that temperature dependent PALS measurements can also be used to probe Tg of water even when the confinement is significantly less severe, i.e. when confined in the pores of Vycor glass, SBA-15 and KIT-6 silicas.
Acknowledgements
This research was financially supported by the Engineering and Physical Sciences Research Council (UK), the National Science and Engineering Research Council (Canada) and the Fonds Qubcois de la Recherche sur la Nature et les Technologies (Province of Quebec).
Footnotes
- 4
For o-Ps to localize in the free volume elements they must persist for at least several nanoseconds.