




Abstract
We show that electron–phonon coupling can induce strong electron pairing in an FeSe monolayer on a SrTiO3 substrate (experimental indications for superconducting  are between 65 and 109 K). The role of the SrTiO3 substrate in increasing the coupling is two-fold. First, the interaction of the FeSe and TiO2 terminated face of SrTiO3 prevents the FeSe monolayer from undergoing a shear-type (orthorhombic, nematic) structural phase transition. Second, the substrate allows an anti-ferromagnetic ground state of FeSe which opens electron–phonon coupling channels within the monolayer that are prevented by symmetry in the non-magnetic phase. The spectral function for the electron–phonon coupling (
are between 65 and 109 K). The role of the SrTiO3 substrate in increasing the coupling is two-fold. First, the interaction of the FeSe and TiO2 terminated face of SrTiO3 prevents the FeSe monolayer from undergoing a shear-type (orthorhombic, nematic) structural phase transition. Second, the substrate allows an anti-ferromagnetic ground state of FeSe which opens electron–phonon coupling channels within the monolayer that are prevented by symmetry in the non-magnetic phase. The spectral function for the electron–phonon coupling ( ) in our calculations agrees well with inelastic tunneling data.
) in our calculations agrees well with inelastic tunneling data.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Small variations of external perturbations can result in the favoring of one of a range of competing structural, electronic, and magnetic ground states for FeSe. In particular, the superconducting transition temperature in FeSe is reputed to vary from almost 0 K when slightly Fe doped [1] to 65 K when placed in a monolayer form on a SrTiO3 substrate [2–5], and transport measurements from a recent work [6] indicate an even larger  , close to 109 K. Although FeSe has a simpler structure to the other iron-based superconductors it resembles components of their structure, and there is the possibility that the mechanism responsible for high temperature superconductivity in monolayer FeSe may extend to other iron-based compounds.
, close to 109 K. Although FeSe has a simpler structure to the other iron-based superconductors it resembles components of their structure, and there is the possibility that the mechanism responsible for high temperature superconductivity in monolayer FeSe may extend to other iron-based compounds.
Early calculations [7, 8] based on density functional theory (DFT) estimated electron–phonon coupling in the iron-based superconductors to be at least 5–6 times too small to explain the transition temperatures found experimentally. Therefore, a large part of the theoretical and the experimental [9–11] work on iron-based superconductors in the literature focused on alternative electron pairing mechanisms such as those associated with magnetic fluctuations. In this letter we suggest that the early first-principle calculations may have underestimated the electron–phonon coupling in FeSe, and we conclude that conventional electron–phonon coupling may be strong enough to contribute significantly to the electron pairing in an FeSe monolayer on SrTiO3 and perhaps other iron-based superconductors.
We focus here on an FeSe monolayer on a TiO2 terminated SrTiO3 substrate. We show that the interaction between the substrate and the FeSe monolayer leads to a high phonon-mediated superconducting  by providing a structural template which holds FeSe near its structural and magnetic phase transitions. When this structural template is not present (as in bulk FeSe or a monolayer of FeSe on a weakly interacting substrate) the system condenses to a different ground state (orthorhombic and non-magnetic) with a reduced electron–phonon coupling.
by providing a structural template which holds FeSe near its structural and magnetic phase transitions. When this structural template is not present (as in bulk FeSe or a monolayer of FeSe on a weakly interacting substrate) the system condenses to a different ground state (orthorhombic and non-magnetic) with a reduced electron–phonon coupling.
Among the many possible ground states of FeSe, calculations based on a semi-local density approximation (GGA) to the DFT select a ground state inconsistent with structural [12], electronic [13–16], and magnetic [12, 17] measurements. While the shortcomings of standard GGA bands for transition metals (such as Fe) can often be corrected by semi-empirically including a Hubbard or a Hund interaction (as in the GGA+U method, [18]), this is not the case for FeSe [19]. Higher levels of theory, such as GW or DMFT in [20, 21], can correctly reproduce most electronic properties of bulk FeSe; however, calculation of the electron–phonon coupling with these methods relies on a simplified deformation potential approximation, as in [22] since electron–phonon coupling matrix elements are difficult to obtain.
Here we show that making the potential on the iron atoms slightly more repulsive for electrons renormalizes the bands near the Fermi level and selects a ground state of FeSe consistent with most experimental data. More specifically, in this method (GGA+A), we empirically1
replace the potential  within the semi-local density approximation (GGA) with
within the semi-local density approximation (GGA) with
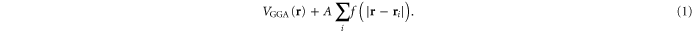
The idea here is to mitigate empirically the fact that the GGA exchange-correlation potential is not the self energy without the second term in equation (1). We find that the detailed form of the dimensionless function  is irrelevant for the computed physical properties of FeSe, as long as f(r) is peaked on the Fe atom (placed at
is irrelevant for the computed physical properties of FeSe, as long as f(r) is peaked on the Fe atom (placed at  ) and the extent of f(r) is comparable with the size of the iron atom d-orbital2
. Next, for a fixed f(r), we tune the parameter A from 0 up to
) and the extent of f(r) is comparable with the size of the iron atom d-orbital2
. Next, for a fixed f(r), we tune the parameter A from 0 up to  (
( ) until3
one of the properties of FeSe (here, occupied bandwidth of the M-point electron pocket) agrees with experimental data (compare blue and green curves in figure 1). Remarkably, using
) until3
one of the properties of FeSe (here, occupied bandwidth of the M-point electron pocket) agrees with experimental data (compare blue and green curves in figure 1). Remarkably, using  improves other salient properties of FeSe as well. For example, the gap (
improves other salient properties of FeSe as well. For example, the gap ( in table 1) at the bottom of the M pocket, and the energy of the Γ band just below the Fermi level are improved in the GGA+A, as well as the peak positions in the density of states at 4 and 6 eV below the Fermi level4
. Magnetic properties are improved as well. Using the experimental crystal structure from [25] in both cases, the GGA+A predicts bulk FeSe to be nonmagnetic as in experiment, while GGA predicts large antiferromagnetically aligned magnetic moments μ on the iron atoms (favored by 0.5 eV per two Fe atoms over the non-magnetic ground state). Finally, the crystal structure is improved in the GGA+A case. A slight shear present in the experimental structure as in [1] (
in table 1) at the bottom of the M pocket, and the energy of the Γ band just below the Fermi level are improved in the GGA+A, as well as the peak positions in the density of states at 4 and 6 eV below the Fermi level4
. Magnetic properties are improved as well. Using the experimental crystal structure from [25] in both cases, the GGA+A predicts bulk FeSe to be nonmagnetic as in experiment, while GGA predicts large antiferromagnetically aligned magnetic moments μ on the iron atoms (favored by 0.5 eV per two Fe atoms over the non-magnetic ground state). Finally, the crystal structure is improved in the GGA+A case. A slight shear present in the experimental structure as in [1] ( ) remains in the GGA+A approach after the structural relaxation, while it disappears in the GGA calculations (
) remains in the GGA+A approach after the structural relaxation, while it disappears in the GGA calculations ( ).
).

Figure 1. Electronic band structure near the M point (full band structure is shown in the supplement (see footnote 5) of monolayer FeSe on SrTiO3 in GGA (red), GGA+A with  (blue), and experimental results. We fit the different experimental data to a parabola (light [3], medium [4], and dark green [5]).
(blue), and experimental results. We fit the different experimental data to a parabola (light [3], medium [4], and dark green [5]).
Download figure:
Standard image High-resolution imageTable 1.
A comparison of the magnetic moment on the iron atom (μ), shear angle α (measured between the primitive unit cell vectors a and b), top of the Γ band (EΓ) and bottom of the M band (occupied bandwidth, EM) relative to the Fermi level, and the band splitting at the M point ( ) in GGA, GGA+A using
) in GGA, GGA+A using  , and from experiments [1, 3–5, 17]. Parameter A is tuned to A = Ac so that occupied bandwidth of the M-point electron pocket (EM) agrees with experimental data. However, using A = Ac significantly improves other properties of FeSe as well.
, and from experiments [1, 3–5, 17]. Parameter A is tuned to A = Ac so that occupied bandwidth of the M-point electron pocket (EM) agrees with experimental data. However, using A = Ac significantly improves other properties of FeSe as well.
| Bulk | Monolayer on SrTiO3 | |||||
|---|---|---|---|---|---|---|
| μ a | α b | μ |
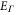
|
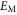
|
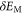
|
|
(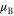 ) ) |
(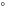 ) ) |
(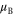 ) ) |
(eV) | (eV) | (eV) | |
| GGA | 2.4 | 90 | 2.6 | 0.66 | 0.19 | 0.02 |
GGA+A (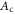 ) ) |
0 | 89.96 | 1.8 | 0.17 | 0.07 | 0.06 |
| Experiment | 0 | 89.7 | c | 0.08 | 0.06 | 0.05 |
aUsing experimental crystal structure. bFully relaxed with the van der Waals correction from [26]. cUnknown.
In these and subsequent calculations we fixed the doping of FeSe monolayer to the level of 0.09 electrons per one Fe atom (as found in ARPES experiments). In the experiment, this doping likely occurs due to presence of oxygen vacancies in the SrTiO3 substrate.
Our focus here is on the electron–phonon coupling and superconductivity in monolayer FeSe. The underlying origin of the success of the GGA+A is an interesting open question and is left for future studies. We only note here two points in favor of GGA+A. First, portion of the electron self-energy  that is missing in the semi-local density approximation is typically large only when
that is missing in the semi-local density approximation is typically large only when  is comparable to the bond length[27], just as for the case of the form of f(r). Second, agreement between GGA+A and experiment is improved not only in monolayer FeSe studied here, but also in bulk KCuF3, LaNiO3, (La,Sr)2CuO4, SrTiO3 (see supplement5
), and (Ba,K)Fe2As2 [28].
is comparable to the bond length[27], just as for the case of the form of f(r). Second, agreement between GGA+A and experiment is improved not only in monolayer FeSe studied here, but also in bulk KCuF3, LaNiO3, (La,Sr)2CuO4, SrTiO3 (see supplement5
), and (Ba,K)Fe2As2 [28].
Equipped with a better FeSe band structure and ground state than obtained from a standard GGA calculation, we are now in a position to compute the electron–phonon coupling strength in the FeSe monolayer. First we discuss the crystal structure of FeSe used in the electron–phonon calculation. Bulk FeSe consists of stacked, weakly interacting, layers of FeSe. Below 90 K these layers are observed to be slightly sheared as shown in figure 2(a) and discussed in [1] (shear is also present in GGA+A calculation, but not in GGA). This shear (nematic) distortion is conventionally described as primitive-tetragonal to base-centered-orthorhombic structural phase transition.

Figure 2. Exaggerated structural distortions in FeSe bulk, an epitaxially constrained monolayer, and a monolayer on SrTiO3. Small circles are Fe atoms and large circles are Se atoms. Primitive unit cell is shown with a dashed gray line.
Download figure:
Standard image High-resolution imageSince the FeSe layers in bulk are only weakly interacting, we expect that the tendency towards a shear distortion will be present even in an isolated single layer of FeSe. This is indeed what we find in the case of monolayer FeSe. Even if we epitaxially constrain the isolated monolayer FeSe unit cell to a cubic SrTiO3 lattice, it still undergoes a local shear-like structural transition shown in figure 2(b) (again, only in GGA+A, not in GGA).
However, once FeSe is placed on a TiO2 terminated SrTiO3 substrate, we find that the interaction of Ti and Se atoms together with the epitaxial strain is able to stabilize FeSe to a nearly square arrangement (see figure 2(c) and supplement (see footnote 5)). A small remnant of the structural distortion present in FeSe is responsible for the electronic gap ( ) at the M point shown in figure 1 and in table 1. (An additional smaller component of the gap results from a built-in electric field between FeSe and SrTiO3, as discussed in [19].) In addition, in the FeSe monolayer on SrTiO3, an antiferromagnetic checkerboard ground state is preferred by 0.11 eV (per unit cell with two Fe atoms) within GGA+A over the non-magnetic one, despite the fact that the opposite is the case for bulk FeSe.
) at the M point shown in figure 1 and in table 1. (An additional smaller component of the gap results from a built-in electric field between FeSe and SrTiO3, as discussed in [19].) In addition, in the FeSe monolayer on SrTiO3, an antiferromagnetic checkerboard ground state is preferred by 0.11 eV (per unit cell with two Fe atoms) within GGA+A over the non-magnetic one, despite the fact that the opposite is the case for bulk FeSe.
The main effect of the SrTiO3 on the FeSe is the structural stabilization described above of a non-sheared and antiferromagnetic ground state. Selection of this ground state then affects the electronic and magnetic properties of FeSe, but only indirectly through the fact that FeSe is in this particular state. The direct effect of the SrTiO3 on the electronic structure of an FeSe monolayer near the Fermi level is negligible. For example, relaxing the structure of FeSe on SrTiO3 and then removing SrTiO3 atoms from the calculation does not affect the electronic structure near the Fermi level (see figure 1 in the supplement (see footnote 5)). Therefore to speed up the calculation of the electron–phonon coupling, we perform calculations on an isolated FeSe layer, without explicitly including SrTiO3. To avoid the shear instability in the FeSe monolayer from removing of SrTiO3, we reduce the value of parameter A in equation (1) from  to
to  and confirm that the electron–phonon matrix elements are not affected by this simplification by carrying out full calculation (see table 1 in the supplement (see footnote 5)).
and confirm that the electron–phonon matrix elements are not affected by this simplification by carrying out full calculation (see table 1 in the supplement (see footnote 5)).
We use state-of-the-art Wannier interpolation technique from [29] and the Quantum-Espresso package described in [30] to calculate the electron–phonon coupling in the FeSe monolayer with a very fine grid in the Brillouin zone (40 × 40). We obtained the superconducting transition temperature  by solving the Eliashberg equation [31, 32] as described in [33]. Figure 3 shows the calculated Eliashberg spectral function
by solving the Eliashberg equation [31, 32] as described in [33]. Figure 3 shows the calculated Eliashberg spectral function  of the FeSe monolayer. We focus our analysis on two groups of phonons for which the electron–phonon coupling is the largest. The first group of phonons (labeled 1 in figure 3) corresponds to phonons with frequency close to 10 meV, and the second group (labeled 2) to phonons with 20 meV (in GGA those frequencies are 15 and 25 meV, respectively).
of the FeSe monolayer. We focus our analysis on two groups of phonons for which the electron–phonon coupling is the largest. The first group of phonons (labeled 1 in figure 3) corresponds to phonons with frequency close to 10 meV, and the second group (labeled 2) to phonons with 20 meV (in GGA those frequencies are 15 and 25 meV, respectively).

Figure 3. Electron–phonon coupling  and phonon density of states
and phonon density of states  (in meV−1) in GGA and GGA+A (using
(in meV−1) in GGA and GGA+A (using  ).
).
Download figure:
Standard image High-resolution imageWhile phonons 1 contribute to about two-thirds of the total electron–phonon coupling strength λ, they contribute to about half of the integrated  spectral function (since they have a lower frequency).
spectral function (since they have a lower frequency).
The atomic displacement character of the two groups of phonons is different. Phonons 1 correspond to a branch of phonons that involve transverse, mostly in-plane displacements of atoms (these phonons cause bulk FeSe to undergo a shear phase transition), while phonons 2 correspond to an out-of-plane transverse displacement of Fe atoms. Furthermore, phonons 1 and 2 couple different parts of the electron Fermi surface at M. Phonons 1 couple mostly at parts of the reciprocal space where the Fermi surface (electron M pocket) crosses the M–Γ line and the least where it crosses the M–X line. The opposite is true for phonons 2. However, since both phonons contribute about equally to  the total electron–phonon coupling (1 and 2 taken together) is nearly constant on the entire M pocket Fermi surface.
the total electron–phonon coupling (1 and 2 taken together) is nearly constant on the entire M pocket Fermi surface.
Hence the importance of the SrTiO3 substrate for increasing the superconducting transition temperature within the phonon mechanism in FeSe is two-fold. First, it prevents phonons 1 from becoming unstable and induce a structural phase transition (as in bulk FeSe). Second, SrTiO3 keeps FeSe in the checkerboard magnetic phase which allows coupling of phonons from groups 1 and 2. In the non-magnetic case, the coupling of these phonons is zero by symmetry [34]. Calculations in [35, 36] also found a significantly smaller electron–phonon coupling in the non-magnetic phase than in the magnetic phase. We also note that at this time, there is no direct experimental measurement of magnetic order in FeSe monolayer on SrTiO3. However, the measured ARPES band structure is most closely resembled to that of the band structure of FeSe with an antiferromagnetic checkerboard order, both in our GGA+A calculation and in previous work [19, 37]. Nevertheless, it is possible that the true ground state of FeSe monolayer consists of fluctuating antiferromagnetic moments on iron atoms. Treatment of electron–phonon coupling in such a state from first-principles goes well beyond the scope of this work.
Comparing  in GGA and GGA+A (figure 3), we find two reasons for an increased coupling in GGA+A. First, preference for a shear distortion in GGA+A increases the electron–phonon matrix elements of phonons 1 (see figure 3 in the supplement (see footnote 5)). Second, the bottom of the electron M pocket
in GGA and GGA+A (figure 3), we find two reasons for an increased coupling in GGA+A. First, preference for a shear distortion in GGA+A increases the electron–phonon matrix elements of phonons 1 (see figure 3 in the supplement (see footnote 5)). Second, the bottom of the electron M pocket  is closer to the Fermi level in GGA+A than in the GGA. Therefore, owing to this band renormalization (narrowing of the occupied bandwidth), the density of states at the Fermi level in GGA+A is larger than in GGA (see table 2 here and figure 3 in the supplement (see footnote 5)). Since λ is proportional to the density of states, it is therefore increased in GGA+A.
is closer to the Fermi level in GGA+A than in the GGA. Therefore, owing to this band renormalization (narrowing of the occupied bandwidth), the density of states at the Fermi level in GGA+A is larger than in GGA (see table 2 here and figure 3 in the supplement (see footnote 5)). Since λ is proportional to the density of states, it is therefore increased in GGA+A.
However, as discussed earlier, we calculated the electron–phonon coupling λ within GGA+A with a reduced value of parameter A from equation (1). Taking into account calculated density of states (1.5 eV−1) with  and
and  (1.8 eV−1) we conservatively estimate that the value of λ at
(1.8 eV−1) we conservatively estimate that the value of λ at  is
is  . Next we use the Eliashberg theory and obtain a conservative estimate of the superconducting transition temperature
. Next we use the Eliashberg theory and obtain a conservative estimate of the superconducting transition temperature  of 26 K (with
of 26 K (with  ) and 21 K (with
) and 21 K (with  ). This estimate is significantly closer to experiment than a standard GGA result (0.1–1.5 K).
). This estimate is significantly closer to experiment than a standard GGA result (0.1–1.5 K).
This range of estimated transition temperatures (21–26 K) is close to the value found across the families of bulk iron-based superconductors. Now we discuss possible reasons for an even larger  in the case of an FeSe monolayer on SrTiO3 (65–109 K).
in the case of an FeSe monolayer on SrTiO3 (65–109 K).
When λ is large, transition temperature is proportional to [38]
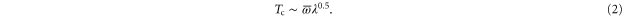
Here  is the averaged phonon frequency and λ is the Brillouin zone averaged electron–phonon coupling strength. Therefore one possibility to get larger
is the averaged phonon frequency and λ is the Brillouin zone averaged electron–phonon coupling strength. Therefore one possibility to get larger  is to further increase λ. It is at least plausible that this could happen for phonons 1, since their contribution to λ is increased when FeSe is approaching the shear-like structural phase transition.
is to further increase λ. It is at least plausible that this could happen for phonons 1, since their contribution to λ is increased when FeSe is approaching the shear-like structural phase transition.
The second possibility is to increase the average frequency  by pairing electrons with high frequency modes (phonons or some other bosons) in addition to phonons 1 and 2. One possibility are magnetic fluctuations [8]. The role of magnetism for superconductivity in FeSe is additionally enriched by the fact that, in the nonmagnetic phase, certain electron–phonon interaction channels are forbidden by symmetry. In addition, structural and magnetic order parameters are strongly coupled in FeSe. For example, bulk orthorhombic FeSe prefers a non-magnetic state, while a cubic FeSe monolayer on SrTiO3 prefers an antiferromagnetic state.
by pairing electrons with high frequency modes (phonons or some other bosons) in addition to phonons 1 and 2. One possibility are magnetic fluctuations [8]. The role of magnetism for superconductivity in FeSe is additionally enriched by the fact that, in the nonmagnetic phase, certain electron–phonon interaction channels are forbidden by symmetry. In addition, structural and magnetic order parameters are strongly coupled in FeSe. For example, bulk orthorhombic FeSe prefers a non-magnetic state, while a cubic FeSe monolayer on SrTiO3 prefers an antiferromagnetic state.
Another tempting possibility suggested in [3] is to pair FeSe electrons to a high-frequency (80 meV) phonon in the SrTiO3 substrate. This coupling was experimentally determined to be large near the origin of the phonon Brillouin zone ( ). Adding experimentally estimated values of the electron–phonon coupling from [3] to our calculated
). Adding experimentally estimated values of the electron–phonon coupling from [3] to our calculated  increases the estimated superconducting transition temperature to 47 K (assuming
increases the estimated superconducting transition temperature to 47 K (assuming  ), even closer to the experimentally determined value (65–109 K).
), even closer to the experimentally determined value (65–109 K).
In closing, we note that the experimentally inferred superconducting  is nearly the same for an FeSe monolayer on TiO2 terminated SrTiO3 [2, 6], BaTiO3 [39], as well as 2% strained SrTiO3 [40]. This observation is consistent with our structural stabilization mechanism since in all three cases interaction between Ti atoms in the TiO2 layer and Se atoms in FeSe is likely the same. However, when a FeSe monolayer is placed on a substrate with a different bonding environment, such as SiC in [41–43] the superconducting
is nearly the same for an FeSe monolayer on TiO2 terminated SrTiO3 [2, 6], BaTiO3 [39], as well as 2% strained SrTiO3 [40]. This observation is consistent with our structural stabilization mechanism since in all three cases interaction between Ti atoms in the TiO2 layer and Se atoms in FeSe is likely the same. However, when a FeSe monolayer is placed on a substrate with a different bonding environment, such as SiC in [41–43] the superconducting  is only 2–9 K. Another indication for the importance of structural stabilization comes from [1]. This study found that bulk FeSe doped with only 2% of iron stays tetragonal (non-sheared) even well below 90 K. This loss of preference for shear is accompanied with loss of superconductivity (
is only 2–9 K. Another indication for the importance of structural stabilization comes from [1]. This study found that bulk FeSe doped with only 2% of iron stays tetragonal (non-sheared) even well below 90 K. This loss of preference for shear is accompanied with loss of superconductivity ( K), again consistent with our finding that keeping FeSe close to a shear (orthorhombic, nematic) structural phase transition increases the electron–phonon coupling strength. Another indication of contribution from electron–phonon mechanism is described in [44] on iron isotope effect measurement.
K), again consistent with our finding that keeping FeSe close to a shear (orthorhombic, nematic) structural phase transition increases the electron–phonon coupling strength. Another indication of contribution from electron–phonon mechanism is described in [44] on iron isotope effect measurement.
Finally, our calculation is consistent with the inelastic scanning tunneling microscope (STM) measurements from [45] in two respects. First, the superconducting gap in the STM measurements (as well as in ARPES in [4, 40]) is node-less, just as is our calculated electron–phonon coupling being nearly constant around the M pocket. Second, both our calculation and the STM measurements find two peaks in the density of states above the superconducting gap Δ (see figure 4). One of these peaks is at 10 meV and another at 20 meV above the gap. As shown in [46], features in the tunneling spectrum above the gap can be associated with  . Therefore, we tentatively assign the two peaks found in the STM measurements to the strongly electron–phonon coupled modes 1 and 2 discussed earlier in the text.
. Therefore, we tentatively assign the two peaks found in the STM measurements to the strongly electron–phonon coupled modes 1 and 2 discussed earlier in the text.

Figure 4. The density of states within the Eliashberg theory calculated using GGA+A and the STM measurement from [45]. The energy is measured relative to the superconducting gap Δ.
Download figure:
Standard image High-resolution imageAcknowledgments
This research was supported by the Theory Program at the Lawrence Berkeley National Lab through the Office of Basic Energy Sciences, US Department of Energy under Contract No. DE-AC02-05CH11231 which provided for the electron–phonon calculation; and by the National Science Foundation under Grant No. DMR10-1006184 which provided for the structural and magnetic study. This research used resources of the National Energy Research Scientific Computing Center, which is supported by the Office of Science of the US Department of Energy.
Footnotes
- 1
- 2
For example, we tried
 with several choices of , , and B all giving similar results.
with several choices of , , and B all giving similar results. - 3
We give in the supplement (see footnote 5) the numerical value of
, a list of all parameters used in the calculations, and a figure showing the dependence of several physical properties of FeSe monolayer on the value of A. - 4
In GW calculations from [21] these same peaks near 4 and 6 eV were found to agree well with the experiment.
- 5
See supplemental material at stacks.iop.org/NJP/17/073027/mmedia for more details.
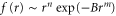

{kind=link}
{kind=link}
{kind=link}
{kind=link}