




Abstract
Ultrafast electronic and structural dynamics of matter govern rate and selectivity of chemical reactions, as well as phase transitions and efficient switching in functional materials. Since x-rays determine electronic and structural properties with elemental, chemical, orbital and magnetic selectivity, short pulse x-ray sources have become central enablers of ultrafast science. Despite of these strengths, ultrafast x-rays have been poor at picking up excited state moieties from the unexcited ones. With time-resolved anti-Stokes resonant x-ray Raman scattering (AS-RXRS) performed at the LCLS, and ab initio theory we establish background free excited state selectivity in addition to the elemental, chemical, orbital and magnetic selectivity of x-rays. This unparalleled selectivity extracts low concentration excited state species along the pathway of photo induced ligand exchange of Fe(CO)5 in ethanol. Conceptually a full theoretical treatment of all accessible insights to excited state dynamics with AS-RXRS with transform-limited x-ray pulses is given—which will be covered experimentally by upcoming transform-limited x-ray sources.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
With the rapid evolution of sub-picosecond and femtosecond x-ray sources and particularly with the emergence x-ray free-electron lasers, first steps to join the unique electronic structure information of x-ray spectroscopy with the ultrafast time scale of dynamics in matter have been taken [1–8]. Often though, time resolved x-ray spectroscopy at these dilute transient species with x-ray absorption, x-ray fluorescence or electron spectroscopy suffer often from the difficult separation between the overlapping spectral signatures of the dilute excited state species and the ground state. To reach improved chemical selectivity with x-ray lasers nonlinear and multidimensional approaches have been explored both theoretically and experimentally [9–15]. In this work, we show how anti-Stokes resonant x-ray Raman scattering (AS-RXRS) adds unique excited state selectivity to already highly selective resonant inelastic x-ray scattering (RIXS) technique. In recent years, RIXS as a resonantly driven Stokes x-ray Raman process has gradually reached relevant energy scales [16–18] to map out magnetic [19], charge [20], orbital [21] and structural excitations [22, 23] as well as complex potential energy surfaces [24, 25] of functional materials and chemical processes. Additionally, stimulated x-ray Raman scattering was recently used to enhance the RIXS signals [26, 27].
To establish AS-RXRS we use the photochemical pathway of the photoinduced ligand exchange reaction of Fe(CO)5 in ethanol solution (figure 1(A)) and conduct AS-RXRS at the Fe L3-edge of the electronically excited Fe(CO)5 and Fe(CO)4 species transiently present along the reaction pathway toward the ligand substituted Fe(CO)4EtOH (figure 1(B)) [28, 29]. This dynamic pathway has been introduced for the gas phase by Trushin et al in [30] and modified for ethanol solution by the mechanism of ultrafast ligand addition and spin crossover by previous work of the authors [28, 29]. Cascading dynamics of electronically excited states in Fe(CO)5 is initiated by resonant absorption of a 266 nm (4.66 eV) photon. The initial photo-absorption creates a metal-to-ligand charge-transfer (MLCT) 1E' state which due to Jahn–Teller-like nuclear dynamics converts through possibly multiple internal conversions to a ligand field (LF) 1E' state with a time constant of 21 fs [30]. The conversion therefore includes a relaxation of an electron from delocalized 2π* orbital to localized  orbital (electron back-transfer). Since
orbital (electron back-transfer). Since  is strongly σ-antibonding with respect to the Fe-CO bond, a Jahn–Teller-like motion on the 1E'(LF) surface leads to a transition state (15 fs) which is followed by a dissociation of a single CO ligand and the creation of Fe(CO)4 in an excited 1B2(LF) state (30 fs) [30]. This electronically excited state evolution we summarize as 1E'(MLCT) → 1E'(LF) and 1E'(LF) → 1B2(LF), with 20 fs and 45 fs time constants, respectively (figure 1(B)). Later, ultrafast ligand addition, spin crossover and geminate recombination finally leads to a branching from the vibrationally hot Fe(CO)4 1A1 that is in the electronic ground state to the ground state ligand substituted complex Fe(CO)4EtOH and the picosecond lived high spin Fe(CO)4 3B2 state [30]. The electronically excited states involved (1E'(MLCT), 1E'(LF) and 1B2(LF)) can lead in resonant x-ray Raman Scattering to the emission of x-ray photons with an energy higher than the incident x-ray photon energy (hυX,out > hυX,in), which is the anti-Stokes Raman signature in this x-ray analog to optical time-resolved resonant Raman spectroscopy. As depicted in figure 1(C) this AS-RXRS energy transfer/anti-Stokes shift corresponds to the valence electronic excitation energies.
is strongly σ-antibonding with respect to the Fe-CO bond, a Jahn–Teller-like motion on the 1E'(LF) surface leads to a transition state (15 fs) which is followed by a dissociation of a single CO ligand and the creation of Fe(CO)4 in an excited 1B2(LF) state (30 fs) [30]. This electronically excited state evolution we summarize as 1E'(MLCT) → 1E'(LF) and 1E'(LF) → 1B2(LF), with 20 fs and 45 fs time constants, respectively (figure 1(B)). Later, ultrafast ligand addition, spin crossover and geminate recombination finally leads to a branching from the vibrationally hot Fe(CO)4 1A1 that is in the electronic ground state to the ground state ligand substituted complex Fe(CO)4EtOH and the picosecond lived high spin Fe(CO)4 3B2 state [30]. The electronically excited states involved (1E'(MLCT), 1E'(LF) and 1B2(LF)) can lead in resonant x-ray Raman Scattering to the emission of x-ray photons with an energy higher than the incident x-ray photon energy (hυX,out > hυX,in), which is the anti-Stokes Raman signature in this x-ray analog to optical time-resolved resonant Raman spectroscopy. As depicted in figure 1(C) this AS-RXRS energy transfer/anti-Stokes shift corresponds to the valence electronic excitation energies.
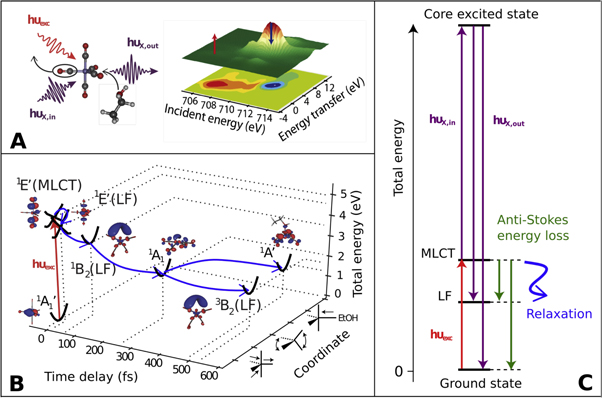
Figure 1. Photo-induced ligand substitution of Fe(CO)5 in ethanol solution into Fe(CO)4EtOH and its relation to AS-RXRS. (A) Depiction of ligand substitution and AS-RXRS at the Fe L3 edge. (B) Reaction pathway involving the electronic ground states of Fe(CO)5  Fe(CO)4 1A1, Fe(CO)4 3B2, Fe(CO)4EtOH 1A' and the electronically excited states of Fe(CO)5 1E' (MLCT), Fe(CO)5 1E'(LF), and Fe(CO)4 1B2 (LF), based on [28, 29] that are in this work selectively detected by AS-RXRS. For each state the molecular orbital occupied by the active (highest energy) electron is shown. (C) Mechanism of AS-RXRS in a total energy picture.
Fe(CO)4 1A1, Fe(CO)4 3B2, Fe(CO)4EtOH 1A' and the electronically excited states of Fe(CO)5 1E' (MLCT), Fe(CO)5 1E'(LF), and Fe(CO)4 1B2 (LF), based on [28, 29] that are in this work selectively detected by AS-RXRS. For each state the molecular orbital occupied by the active (highest energy) electron is shown. (C) Mechanism of AS-RXRS in a total energy picture.
Download figure:
Standard image High-resolution imageResults
In figure 2 we show on the left side panels the electronic orbital structure of ground state Fe(CO)5 (figure 2(A)) and how the Stokes resonant x-ray Raman scattering (also commonly denoted as resonant inelastic x-ray scattering, RIXS) leads in the schematic representation of the inelastic x-ray scattering plane (figure 2(C)) to a participator channel with zero energy transfer, and to Stokes energy transfer (loss) due to the creation of final state electron–hole pairs. In figure 2(E) the RIXS map of the Fe(CO)5 ground state calculated with ab initio restricted active space self-consistent field (RASSCF) method [31] is shown (see methods for further details). On the right-hand side panels (B) and (D) of figure 2, we describe how in an one-electron orbital picture the creation of an initial electronic MLCT excitation through optical absorption leads to the opening of a lower energy scattering resonance and the occurrence of fully separated AS-RXRS spectral signatures. Different from the ground state, the optically excited Fe(CO)5 1E'(MLCT) state has a valence vacancy in the dπ orbital. Thus a new energetically lower x-ray scattering resonance is opened up, depicted in figure 2(D). Resonant x-ray Raman Scattering through this excited state resonance occurs at a hvexc-red-shifted core-level resonance energy and leads for all electron–hole pair final states to the appearance of inelastic scattering features with hvexc-blue-shifted emission energies in relation to the ground state situation. This is the signature of AS-RXRS. The Resonant x-ray Raman Scattering planes calculated using the RASSCF method in the ground state and in the 1E'(MLCT) excited state in figures 2(E) and (F) confirm the conceptual reasoning based on the simplified one-electron orbitals. Therefore the AS-RXRS spectral features are a result of excitation to the dπ vacancy that is followed by a decay of the excited electrons at the 2π* orbital. The most intense AS-RXRS peak in figure 2(F) is at −5 eV energy transfer which corresponds to scattering to the  ground state. Dipole selection rules apply to all the involved transitions (pump and probe) and we note that in case of molecules with inversion symmetry, anti-Stokes scattering from optically populated state to the ground state is dipole forbidden. This is not the case for Fe(CO)5 which belongs to the D3h point group symmetry and thus has no inversion center.
ground state. Dipole selection rules apply to all the involved transitions (pump and probe) and we note that in case of molecules with inversion symmetry, anti-Stokes scattering from optically populated state to the ground state is dipole forbidden. This is not the case for Fe(CO)5 which belongs to the D3h point group symmetry and thus has no inversion center.

Figure 2. Excited state selectivity of anti-Stokes resonant Raman scattering left column; electronic ground state, right column; additional optically created MLCT excitation. (A) Molecular orbital structure of Fe(CO)5 showing relevant orbitals for Fe L3-edge RIXS and one-electron transitions through 2π* core resonance for the electronic ground state (GS). (B) One-electron transitions involved in the pump-probe anti-Stokes scattering of the optically populated 1E'[MLCT, (dπ)−1(2π*)1] excited state. (C) Schematic Fe(CO)5 ground state RIXS map at the 2π* resonance (711.5 eV) based on the MO structure of the complex (dσ* resonance is omitted for clarity). (D) Schematic RIXS map of the (dπ)−1(2π*)1 MLCT excited state showing scattering through 2π* resonance (711.5 eV) and through dπ hole (706.5 eV) which leads to anti-Stokes scattering (peak at −5 eV energy transfer). (E) Calculated RIXS map of the ground state and (F) the 1E'[MLCT, (dπ)−1(2π*)1] excited state. (5σ)−1(dσ*)1 and (5σ)−1(2π*)1 final states were not included in the calculations. Labels next to scattering features show the final state.
Download figure:
Standard image High-resolution imageIn figure 3 we show all AS-RXRS maps of the excited states along the Fe(CO)5 photodissociation pathway. The projections through the scattering planes (white dashed regions) in the AS-RXRS region yield the computed AS-RXRS matching the experimental ones. In particular, we can define spectral regions R1, R2, R3 (black solid regions) that detect background-free the AS-RXRS features of electronically excited states 1E'(MLCT) Fe(CO)5, 1E'(LF) Fe(CO)5 and 1B2(LF) Fe(CO)4, respectively. In addition (R4) picks up the 3B2(LF) Fe(CO)4 electronic ground state reached after CO detachment. Transient occupation of these states is shown on the right-hand side panels of figure 3. We note that computational treatment of AS-RXRS from different excited molecules have been done before by Tanaka et al and Pandey et al [32, 33].
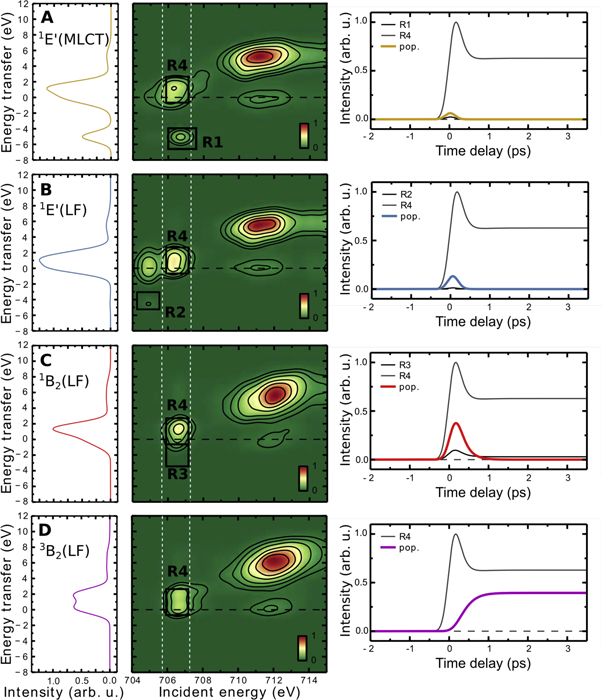
Figure 3. Computed RASSCF resonant inelastic x-ray scattering maps (center column) of the excited states (A) 1E'(MLCT) Fe(CO)5; (B) 1E'(LF) Fe(CO)5; (C) 1B2(LF) Fe(CO)4 and ground state photoproduct (D) 3B2(LF) Fe(CO)4 using the experimental resolution. Left column: resonant inelastic x-ray scattering spectra extracted for 706–707 eV incident photon energy. Right column shows population dynamics of the respective state (colored lines) and the AS-RXRS intensities of regions R1, R2, R3 selective to the respective excited states (R1) 1E'(MLCT) Fe(CO)5; (R2) 1E'(LF) Fe(CO)5; (R3) 1B2(LF) Fe(CO)4 as well as region R4 picking up the 3B2(LF) ground state of Fe(CO)4.
Download figure:
Standard image High-resolution imageIn figure 4 direct comparison of experimental and simulated Fe L3-edge AS-RXRS signatures along the Fe(CO)5 to Fe(CO)4EtOH ligand substitution pathway is shown. In the panel (A) of figure 4 the resonant x-ray scattering difference map is shown in the center. Along the axis of incident photon energy we observe the opening of the red-shifted x-ray scattering resonance due to the optically created initial 1E'(MLCT) state at −4.6 eV excitation energy equal to the pump photon energy (266 nm). Within this spectral region along the emission energy axis the occurrence of blue-shifted anti-Stokes features occurs. The AS-RXRS region R3 shows the transience of the convoluted temporal resolution and the excited state dynamics. The anti-Stokes intensity disappears after 400 fs, with a deconvoluted exponential time constant of ∼100 fs, visible from the delay scans in figure 4(A). Region R4 of the 3B2 Fe(CO)4 photoproduct retains significant intensity.

Figure 4. Experimental observation and simulations of AS-RXRS at the Fe L3-edge the Fe(CO)5 photochemistry. (A) Experiment. Center: pumped–unpumped difference map containing resonant inelastic x-ray scattering and AS-RXRS; top: projection into x-ray absorption spectra: ground state (black line), at time delay 0.1–0.4 ps (red line) and 0.4–3.6 ps (blue line); left: occurrence of AS-RXRS features between −5 and −1 eV energy transfer at 706–707 eV incident energy, that is present at 0.1–0.4 ps time delay; right: temporal evolution of the intensity in AS-RXRS regions showing excited state dynamics. (B) Simulation with RAS-SCF and the kinetic rate model. Center: pumped–unpumped difference; left: difference spectra at 706–707 eV incident energy at 0.1–0.4 ps and 0.4–3.6 ps, inset shows magnification of an AS-RXRS feature corresponding to E'(MLCT) Fe(CO)5 state; right: delay scans from the respective regions.
Download figure:
Standard image High-resolution imageTo fully describe the experimental observables (figure 4(A)) through the photochemical pathway of figure 1(B) [28–30], we simulate the observed AS-RXRS features using the RASSCF calculations for all involved excited states from figure 3 and the kinetic rate model of the photochemical pathway (figure 1(B)), taking into account the experimental time resolution of 300 fs and the experimental spectral broadening contributions (figure 4(B), and methods). At these conditions the AS-RXRS feature is dominated by the electronically excited 1B2(LF) state of Fe(CO)4 in region R3 (figure 3(C)). The 1E'(MLCT) and 1E'(LF) states (with 20 fs and 45 fs lifetimes, respectively) are less pronounced (inset in the simulated RIXS spectrum in figure 4(B)). The 1B2(LF) Fe(CO)4 state correlates with the 1E'(LF) Fe(CO)5 state, however it has significantly lower energy due to strong structural relaxation which has taken place (i.e. CO dissociation): based on the RASSCF calculation, the 1E'(LF) state is 4.6 eV above the Fe(CO)5 ground state, whereas the relaxed 1B2(LF) state has only ∼1 eV higher energy compared to the Fe(CO)4 1A1 state. Thus this energy relaxation leads to the AS-RXRS feature with a smaller blue shift in comparison to the initial optical excitation energy of hvexc = 4.66 eV. Simulation of the AS-RXRS feature at 706–707 eV incident photon energy region in figure 4(B) reproduces remarkably well the experimental spectral shape (note that RASSCF calculations at the 2π* core resonance at 710–714 eV are less accurate). The lifetime of 1B2(LF) state is ∼100 fs and it relaxes via two parallel process to the 3B2(LF) ground state of four-coordinated Fe(CO)4 or to the 1A1 ground state of five-coordinated Fe(CO)4L (L = EtOH, CO) [21]. This results in disappearance of the anti-Stokes scattering feature (region R3), whereas considerably intensity close to elastic peak at 706–707 eV (region R4) remains due to the 3B2(LF) Fe(CO)4 state (figure 3(D)).
Discussion
We can now demonstrate in figure 5 the full potential of AS-RXRS with transform-limited Gaussian x-ray pulses (ΔEΔt = 0.44 h) from upcoming x-ray lasers [34, 35]. Three prototypical scenarios focusing on the initial electron back-transfer between the Fe(CO)5 1E'(MLCT) and 1E'(LF) states and subsequent CO removal during 1E'(LF) to 1B2(LF) inter conversion are presented. As defined in figure 3, anti-Stokes features of the 1E'(MLCT), 1E'(LF) and 1B2(LF) states are picked up within R1, R2 and R3, located at −5 eV, −4.6 eV and −1 eV energy transfer, respectively. AS-RXRS preserves the bandwidth ΔE of the scattered radiation at linear dispersion with an upper limit given by the natural core-hole lifetime broadening Γ (0.3 eV at the Fe L3-edge) reflecting the Fe L3-edge 2.2 fs natural core-hole lifetime. We give the overall temporal resolution in the simulation as the convolution of probe and pump pulses at delay t (assumed equal in duration). With different pulse length Δt, both the temporal resolution and the chemical selectivity can be varied and finest details of the dynamics and potential energy surfaces of excited states can be extracted background free. Column A of figure 5 shows highest temporal selectivity with a pulse duration of Δt = 1 fs that separates the 1E'(MLCT) and 1E'(LF) states in time, but with ΔE = 2.0 eV incident bandwidth at the cost of no spectral selectivity. However, the features from the 1E'(MLCT) and 1E'(LF) states can still be well distinguished since increasing the bandwidth beyond the core-hole lifetime broadening does not result in further broadening of the spectra. Column B of figure 5 shows optimized chemical selectivity and temporal resolution through a transform limited pulse with ΔE = 0.2 eV and a pulse duration of Δt = 10 fs. This yields distinction both energetically and temporally and separates AS-RXRS features of the 1E'(MLCT), 1E'(LF) and 1B2(LF) states. Column C of figure 5 shows how sub-natural linewidth resolution maps out the potential energy surfaces [22, 25, 36] of the 1E'(MLCT), 1E'(LF) excited states individually for transform limited pulses with ΔE = 0.02 eV and pulse duration of Δt = 100 fs. Although no direct temporal separation occurs, the chemical shift between the x-ray scattering resonances of the different 1E'(MLCT), 1E'(LF) excited states separates them, thus allowing to map the potential energy surface of the excited states undergoing rapid photochemical dynamics.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. AS-RXRS features computed with Gaussian transform limited x-ray pulses exploring three complementary scenarios at the Fe L3-edge and at time delays t demonstrate the ability to resolve excited state selective ultrafast internal conversion between 1E'(MLCT) and 1E'(LF) states (electron back-transfer) and subsequent CO removal during 1E'(LF) to 1B2(LF) inter conversion (1E'(MLCT), 1E'(LF), 1B2(LF) AS-RXRS regions within R1, R2, R3 respectively). The gray areas in the top panels spans ΔEΔt in relation to the dynamics and energies involved in the initial electron back-transfer between the Fe(CO)5 1E'(MLCT) and 1E'(LF) states. (A) Highest temporal selectivity with Δt = 1 fs and ΔE = 2.0 eV. (B) Temporal and spectral selectivity with ΔE = 0.2 eV and Δt = 10 fs. (C) Potential energy surface mapping with sub-natural linewidth energy resolution ΔE = 0.02 eV and Δt = 100 fs.
Download figure:
Standard image High-resolution image{kind=link}
Methods
Computational details
Theoretical x-ray spectra were derived from RASSCF calculations [37] using the MOLCAS-7 software [38]. For further details see [28, 29]. The geometries were optimized with the CASPT2 method [39] and the TZVP basis set [40] for all atoms. The active space contained 12 electrons in 12 orbitals. Some of the geometries were optimized with density functional theory using the PBE functional [41] and the TZVP basis set.
Following experimental factors contribute to the spectral linewidth in the experiment and are taken into account in the simulation of the experiment: the 0.3 eV core-hole lifetime broadening, 0.5 eV incident x-ray bandwidth, 1 eV spectrometer resolution and the 0.5 eV due to inhomogeneous broadening from solvent environment and vibrational effects (all values FWHM). RIXS spectra were simulated using the Kramers–Heisenberg formula. Spectra were calculated for an ensemble of randomly oriented molecules excited by linearly polarized light and detected in the plane of polarization. Interference effects were excluded.
Experimental details
Experiment was performed at the linac coherent light source (LCLS) soft x-ray materials science (SXR) instrument [42, 43] with the liquid jet endsation [44]. The 1 mol l−1 Fe(CO)5 ethanol solution was photo-excited at 266 nm (4.66 eV). The pump-laser pulse duration amounted to 100 fs (FWHM) and the pulse energy was estimated to ∼5 μJ. With a pump-laser spot size of 100 × 400 μm2, this corresponded to a peak fluence of ∼1.25 × 1011 W cm−2. We found no evidence for multi-photon processes at this fluence of the pump laser.
Fe L3-RIXS intensities were measured by scanning the incident photon energy from 703 to 715 eV. The resolution in the RIXS measurements along the incident-photon energy axis is defined by the excitation bandwidth. This amounted here to 0.5 eV (FWHM) and was determined by the slit size of 150 μm of the SXR monochromator. Incident flux was measured on a shot-by-shot basis using intensity monitor installed after the monochromator [45].
Competing financial interests
The authors declare no competing financial interests.
Acknowledgments
Use of the Linac Coherent Light Source (LCLS), SLAC National Accelerator Laboratory, is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SXR Instrument is funded by a consortium whose membership includes the LCLS, Stanford University through the Stanford Institute for Materials Energy Sciences (SIMES), Lawrence Berkeley National Laboratory (LBNL), University of Hamburg through the BMBF priority program FSP 301, and the Center for Free Electron Laser Science (CFEL). Part of this research was conducted within the Helmholtz Virtual Institute VI 419 'Dynamic Pathways in Multidimensional Landscapes'. MO acknowledges support from the Swedish Research Council, Carl Tryggers Foundation, and Magnus Bergvall Foundation. AF acknowledges funding from the ERC-ADG-2014—Advanced Investigator Grant—no. 669531 EDAX under the Horizon 2020 EU Framework Programme for Research and Innovation. The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at the Swedish National Supercomputer Center (NSC) and the High Performance Computer Center North (HPC2N). MB had financial support by the Volkswagen Stiftung. IR, WQ, SG, MS and ST are grateful to SFB755-DFG and SFB1073-DFG for financial support. WZ, RWH, and KJG acknowledge support through the AMOS program within the Chemical Sciences, Geosciences, and Biosciences Division of the Office of Basic Energy Sciences, Office of Science, US Department of Energy.
Author contributions
The manuscript was written by KK and AF with input from all co-authors. IJ and MO carried out the RASSCF calculations. KK, IR, SS, WQ, MB, SG, MS, DN, WZ, RWH, KJG, WFS, JJT, BK, FH, ST, PW and AF carried out the experiment.
Additional information
Correspondence and requests for materials should be addressed to AF.