
Abstract
Liver cell culture models are attractive in both tissue engineering and for development of assays for drug toxicology research. To retain liver specific cell functions, the use of adequate cell types and culture conditions, such as a 3D orientation of the cells and a proper supply of nutrients and oxygen, are critical. In this article, we show how extracellular matrix mimetic hydrogels can support hepatocyte viability and functionality in a perfused liver-on-a-chip device. A modular hydrogel system based on hyaluronan and poly(ethylene glycol) (HA-PEG), modified with cyclooctyne moieties for bioorthogonal strain-promoted alkyne-azide 1, 3-dipolar cycloaddition (SPAAC), was developed, characterized, and compared for cell compatibility to hydrogels based on agarose and alginate. Hepatoma cells (HepG2) formed spheroids with viable cells in all hydrogels with the highest expression of albumin and urea in alginate hydrogels. By including an excess of cyclooctyne in the HA backbone, azide-modified cell adhesion motifs (linear and cyclic RGD peptides) could be introduced in order to enhance viability and functionality of human induced pluripotent stem cell derived hepatocytes (hiPS-HEPs). In the HA-PEG hydrogels modified with cyclic RGD peptides hiPS-HEPs migrated and grew in 3D and showed an increased viability and higher albumin production compared to when cultured in the other hydrogels. This flexible SPAAC crosslinked hydrogel system enabled fabrication of perfused 3D cell culture of hiPS-HEPs and is a promising material for further development and optimization of liver-on-a-chip devices.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Preservation of functional behaviour in ex vivo cell culture models is essential for successful and effective applications in drug development research. In conventional in vitro models, cells are seeded on two-dimensional (2D) surfaces which can lead to impaired cell function [1, 2]. Recently, miniaturized bioreactors with spatially arranged cells, cultured under dynamic conditions, known as organs-on-chips, have emerged as alternative cell culture models to better mimic the cells in vivo environment [3–5]. The liver is of special interest for such models due to the high attrition rate of hepatotoxic drugs [6]. Several previous publications have described organs-on-chips that can improve the function of hepatocytes and by that potentially better predict the outcome of a drug's impact in the clinical trials [7–10]. Current challenges for a wide adoption of organ-on-a-chip devices in both academic and industrial settings include their demanding hands-on operating procedures, the need to replace the common prototyping material polydimethylsiloxane (PDMS), the ability to implement iPS-derived cells, and to minimize the large dead volume caused by reservoirs and tubing [11].
One approach to enhance and prolong the cellular functions within organs-on-chips is to encapsulate cells within a three-dimensional (3D) hydrogel matrix that replicates the supportive functions of the extracellular matrix (ECM) [12]. The hydrogels should ideally mimic critical features of the native tissue-specific pericellular environment, such as mechanical and structural features, presence of cell adhesion motifs and moieties that can sequester bioactive factors, which in turn affect cell viability, proliferation, differentiation and cell–cell communication [13]. Previous efforts to realize 3D in vitro models have utilized the ability of cells (including immortalized cells, primary cells, and stem cell derived cells) to form spheroids, both as mono- and co-cultures, as constructs for a 3D architecture within bioreactors, microtiter plates, and microfluidic devices [14–16]. Spheroids are formed in environments in which the possibility for cells to attach to their surroundings is limited and instead, by adhering to each other, form aggregates. Spheroid formation can also occur in hydrogels that do not feature any active cell-binding motifs [17]. However, by integrating ECM components, such as laminins, or ECM derived cell adhesion peptide motifs, such as the Arg-Gly-Asp (RGD) peptide, it is possible for cells to attach to and migrate in the material, as opposed to forming spheroids [18, 19]. The intrinsic properties of the hydrogels, irrespectively if they are naturally-derived or fully synthetic, are thus of large concern when encapsulating human-derived cells, such as hepatic cells, and motivates comparative studies to identify hydrogel scaffolds that can optimize cellular functions ex vivo. Although widely used as cell culture scaffolds, ECM materials derived from biological sources such as reconstituted basement membrane preparations from sarcoma cells, often suffer from batch-to-batch variability, potential immunogenicity and presence of biological contaminants, and is also difficult to modify in order to optimize the performance for different cell types and applications [20]. Well-defined ECM mimetic hydrogels based on synthetic or semisynthetic components and with properties that can be tailored for the specific application are hence very attractive. To this end numerous examples of hydrogels based on both biopolymers and synthetic polymers, including hyaluronic acid, alginate, and poly(ethylene glycol) have been developed and explored for 3D cell culture and tissue engineering [13]. Precisely defined molecular components that recapitulate properties of their natural ECM counterparts can be enabled by using fully or partly synthetically modified polymers that incorporates e.g. bioactive ligands such as peptides for cell adhesion, and bioorthogonal crosslinking moieties, i.e. chemical reactants that do not interfere with biochemical reactions or cell-structures [21]. This in turn facilitates development of modular and tunable hydrogels and are potentially better candidates for development of robust and reproducible tissue mimetic cell-based assays [22, 23].
Furthermore, from a scaffold fabrication perspective, the method used to prepare hydrogels must be as gentle as possible for the cells to not impede their viability and functionality. This is especially true when culturing non-proliferating cells such as primary- and stem cell derived hepatocytes. Several hydrogels used as biomaterials for 3D cell cultures require the cells to be seeded in environments that potentially could be harmful for the cells. This can either be before crosslinking, e.g. hydrogels made of reconstituted basement membrane preparations from sarcoma cells and collagen where the material is kept on ice during cell seeding, or during crosslinking such as hydrogels crosslinked by UV-irradiation. Recently, hydrogels fabricated using strain-promoted alkyne-azide 1,3-dipolar cycloaddition (SPAAC) have emerged as a rapid bioorthogonal crosslinking method that does not require precursors, initiators, or catalysts, and therefore minimize the risk of potential toxic effects on cells [24–26]. For instance, Takahashi et al utilized SPAAC to form injectable hydrogels by modifying hyaluronan with azide- and cyclooctyne, respectively [24]. These hydrogels were fully biodegradable and showed a high viability of the murine cell line NIH-3T3 after 2 d. However, as pointed out by the authors, their choice of cyclooctyne-derivative has low water solubility and the lack of specific cell adhesion ligand motifs may render the hydrogel less versatile in certain biomedical applications such as 3D cell culture. Jonker et al addressed these issues by using a much more water soluble cyclooctyne bicyclo[6.1.0]nonyne (BCN) and incorporated cell adhesion peptide motifs (GRGDSG) in azide modified multiarmed poly(ethylene glycol) (PEG) polymers [25]. The hydrogels showed attachment and high viability of HOS cells, NIH-3T3 cells and HeLa cells. The incorporation of the RGD-peptide was critical to achieve high cell viabilities but since the RGD peptide was included in the hydrogel components from start, possibilities to investigate the effects of different types and concentrations of cell adhesion motifs was limited. Optimization of ECM-mimicking 3D cell culture models would consequently benefit from modular hydrogel fabrication strategies that enable tuning of mechanical properties and flexible incorporation of cell adhesion motifs.
In this paper, we introduce a modular and flexible hydrogel (HA-PEG) that is crosslinked via SPAAC using a cyclooctyne-modified hyaluronan (HA-BCN) and a multiarmed azide-modified PEG. The mechanical properties and hydrogelation kinetics can be conveniently tuned by modulating the degree of crosslinking and temperature, respectively, which facilitates optimization of culture conditions. In addition, we compare this hydrogel system with two other commonly used hydrogels for 3D cell culture, based on agarose and alginate, on the performance of encapsulated liver carcinoma cells (HepG2) and pluripotent stem cell derived hepatocytes (hiPS-HEP) in a perfused 3D cell culture device to enable implementation into a liver-on-a-chip platform. Furthermore, we show that it is possible to use excess SPAAC moieties to graft different ligands to the hydrogel components, such as RGD-peptides for promoting cell adhesion, without affecting the mechanical properties of the hydrogel. The effect of including either linear or cyclic RGD peptide cell adhesion motifs in the HA-PEG hydrogels on cell survival and functionality of hiPS-HEP is investigated.
2. Materials and methods
Bacterial fermented sodium hyaluronate (denoted HA, Mw 100–150 kDa) was acquired from Lifecore Biomedical. 8-Arm PEG-Azide (denoted p(N3)8, Mw 10 kDa) was acquired from Creative PEGworks. All other chemicals were acquired from Sigma Aldrich if nothing else is stated.
2.1. HA-BCN synthesis
HA-BCN was synthesized as described by Selegård et al [27]. In short, HA was dissolved in MES buffer (100 mM, pH 7). N-[(1 R,8 S,9 S)-Bicyclo[6.1.0]non-4-yn-9-ylmethyloxycarbonyl]-1,8diamino-3,6-dioxaoctane (BCN-NH2) was dissolved in 5:1 (v/v) acetonitrile:water prior addition of EDC and 1-hydroxybenzotriazole hydrate, followed by mixing with the HA. The carbodiimide crosslinking reaction was allowed to proceed for 24 h, followed by dialysis (MWCO 12–14 kDa, Spectra/Por RC, Spectrum Laboratories Inc.) in acetonitrile:water (1:10 v/v) for 24 h, followed by double distilled water for 3 days. The dialyzed product was lyophilized to a dry powder, yielding HA-BCN with a derivatization degree of approximately 19% based on 1H-NMR (supporting figure S1 is available online at stacks.iop.org/BF/11/015013/mmedia).
2.2. Peptide synthesis
The linear RGD-peptide, denoted linRGD-N3, with the amino acid sequence K(N3)-βA-βA-RGDS was synthesized using standard SPPS protocols. In short, the peptide was synthesized on a rink amide resin (ChemMatrix, 0.47 mmol g−1) using 4 equivalents (eq) of Fmoc-protected amino acids (Iris Biotech GmbH, Germany), 4 eq of HCTU and 8 eq of DIPEA. The cyclic RGD-peptide, denoted cRGD-N3, with the amino acid sequence c(RGDfK)(N3), was synthesized according to a procedure described by McCusker et al [28] with some modifications. Fmoc-Asp-OAll was attached by its side chain to a Wang resin (Novabiochem, 1.13 mmol g−1) using 5 eq of Fmoc-protected amino acid, 5 eq of MSNT and 3.75 eq of Melm in dry DCM. The reaction was allowed to proceed for 1 h under N2 atmosphere. Sequentially, the peptide was elongated with Fmoc-Gly-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Lys(Mtt)-OH and Fmoc-D-Phe-OH in sequential order using 5 eq of amino acids, 5 eq TBTU/HOBt and 10 eq of DIPEA using standard SPPS protocols. After the final amino acid was attached, the C-terminal allyl ester (OAll) was removed using 3 eq Pd(PPh3)4 in CHCl3:AcOH:N-methylmorpholine (17:2:1) under N2 atmosphere for 2 h. The resin was washed with alternating solutions of 30 mM DIPEA in DMF and 20 mM diethyl-dithiocarbamic acid in DMF followed by final Fmoc deprotection. On-bead cyclization was accomplished by treatment with 5 eq of TBTU/HOBt and 10 eq DIPEA for 2 h. In continuation, the Mtt protection group was removed by washing the resin with 1% TFA in DCM. The deprotected lysine residue was then reacted with 10 eq of 3-azidopropionic acid, 10 eq NHS/EDC and 25 eq DIPEA in DCM for 1 h. Both the peptides linRGD-N3 and cRGD-N3 were cleaved from their solid support using TFA:H2O:TIS (95:2.5:2.5) for 2 h before being concentrated and precipitated twice in cold diethylether. The precipitate was purified by RP-HPLC using a C-18 column (Kromatek HiQ-Sil C18HS). Mass identity the peptides were confirmed by MALDI-ToF MS (UltraflexXtreme, Bruker Daltonics) using α-cyano-4-hydroxycinnamic acid as matrix (supporting figure S2).
2.3. Hydrogel preparation
2.3.1. HA-PEG
The constituents of HA-PEG hydrogels, HA-BCN (supporting figure S3(a)) and p(N3)8 (supporting figure S3(b)) respectively, were UV-sterilized (60 kJ cm−2) and dissolved at a concentration of 2.0% (w/v) in PBS or cell culture medium. If nothing else is stated, hydrogels were formed by mixing HA-BCN with p(N3)8 at a BCN:N3 ratio of 10:1. In experiments with 2-azidoethanol, HA-BCN was incubated with 10% (v/v) 2-azidoethanol solutions of different concentrations at 37 °C for 1 h prior addition of p(N3)8. For incorporation of cell adhesion promoting RGD-peptides, HA-BCN was first incubated with either linRGD-N3 or cRGD-N3 (1 μM) at 37 °C for 30 min to allow homogeneous distribution of RGD sequences in the final hydrogel. Furthermore, in cell culture experiments, p(N3)8 was first diluted with 10% (v/total hydrogel volume) cell suspension prior mixing with HA-BCN.
2.3.2. Agarose
Agarose was dissolved in PBS at a concentration of 1.25%, autoclaved at 121 °C for 20 min and maintained in liquid form on a 37 °C heat block until further use. For cell culture experiments, agarose hydrogels were mixed 5:1 (v/v) with cell suspension to obtain a final hydrogel concentration of 1%.
2.3.3. Alginate
Alginate powder was UV-sterilized (60 kJ cm−2) and dissolved at a concentration of 1.25% in 0.9% NaCl. For cell culture experiments, alginate hydrogels were mixed 5:1 (v/v) with cell suspension to obtain a final hydrogel concentration of 1%. The hydrogel was formed by incubation in 0.9% CaCl2 for 10 min.
2.4. Rheology
The viscoelastic properties of the hydrogels were assessed by oscillatory rheology (Discovery HR-2, TA Instruments), and all sample conditions were examined in at least duplicates if nothing else is stated. Gelation kinetics studies were conducted with a stainless steel 20 mm 1° cone-plate geometry at 0.1 strain% and 1 Hz oscillatory frequency. Samples for gelation were prepared in situ on the rheometer and all measurements were started within 3 min after mixing the components. In addition, a solvent trap was used for all gelation experiments to prevent dehydration. Furthermore, fully formed hydrogels (1 h gelation, 24 h incubation in PBS) were examined with (1) amplitude sweeps at 1 Hz oscillatory frequency (supporting figures S4) and (2) frequency sweeps at 0.1 strain% with 1.0%–2.0% (w/v) total polymer concentration. In addition, the dynamic viscosity of the individual hydrogel components HA-BCN and p(N3)8 (2.0% w/v) was assessed by flow rheology using a stainless steel 20 mm 1° cone-plate geometry.
2.5. Scanning electron microscopy
The morphology of fully formed hydrogels was examined with scanning electron microscopy (SEM, Leo Gemini, Zeiss) with an acceleration voltage of 5 kV. Samples were chemically dried using increasing concentrations of ethanol (50%, 70%, 80%, 90%, 100%) and finally 100% hexamethyldisilazane for 10 min each. Samples were sputtered with platinum prior imaging.
2.6. Diffusion studies
The relative release of 5(6)-Carboxyfluorescein (CF, 376 Da) and Cy5-labelled bovine serum albumin (BSA-Cy5, 70 kDa) through diffusion from HA-PEG were measured using fluorescence spectroscopy (Fluoromax 4, Horiba Scientific). HA-PEG hydrogels (2.0% w/v, 50 μl) with CF or BSA-Cy5 (100 μM) were formed in the bottom of a semi-macro cuvette. The cuvette was centrifuged during gelation (1 h) to ensure even distribution. Sequentially, 1 ml PBS-buffer was added to the cuvette and the relative release of CF or BSA-Cy5 was monitored over time. Prior to each measurement the PBS-buffer was mixed to ensure homogeneity. The CF-release was monitored at λem = 515 nm (λex = 492 nm) and BSA-Cy5 was monitored at λem = 665 nm (λex = 650 nm).
2.7. Cell culture
HepG2 cells (HB-8065, ATCC) were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acids (NEAA) and 1% penicillin/streptomycin (PEST). Adherent cells were trypsinized, centrifuged and resuspended either in hydrogels at 1 × 106 viable cells ml−1 or in wells coated with poly-L-lysine (PLL) at 4 × 104 viable cells cm−2. Human hepatocyte-like cells derived from a human induced pluripotent stem cell line (Cellartis® enhanced hiPS-HEP v2 from ChiPSC18, Takara Bio Europe AB, Sweden) were cultured according to the specifications from the manufacturer. Briefly, one vial of cryopreserved cells was thawed in a water bath at 37 °C for approximately 1 min and its content was transferred to thawing medium consisting of InVitroGRO HT medium (BioIVT) supplemented with 0.5% PEST and 5 μM Y-27 632. The cells were resuspended, incubated for 15 min at room temperature, and centrifuged at 100x g for 2 min. The cell pellet was resuspended in plating medium consisting of InVitroGRO CP medium (BioIVT) supplemented with 0.5% PEST, 5 μM Y-27 632 and 1x Cellartis HEP additive (Takara Bio Europe AB), and seeded, either in hydrogels at 5 × 106 viable cells ml−1 for 3D cell culture experiments or in wells coated with Cellartis HEP Coat (Takara Bio Europe AB) at 4 × 105 viable cells cm−2 for 2D experiments and incubated at 37 °C and 5% CO2. The next day, cells were washed twice in Williams Medium E (WME, Life Technologies) supplemented with 0.1% PEST, and finally incubated in Cellartis Enhanced hiPS-HEP Maintenance Medium consisting of WME supplemented with 0.1% PEST, 0.5% DMSO and 1x Cellartis HEP additive. The maintenance medium was changed every second day.
2.8. Liver-on-a-chip setup
For perfusion experiments, commercial devices comprising two wells in series and connected to medium reservoirs via channels (μ-Slide III 3D Perfusion IbiTreat, Ibidi, Germany) were used according to a modified protocol from the manufacturer. Cells were resuspended in cell culture medium and seeded directly in the wells of the device, either in hydrogels for 3D cell culture experiments or on PLL or HEP Coat coated surfaces for 2D cell culture experiments. Cells encapsulated in hydrogels or resuspended in cell culture medium were seeded at a volume of 25 μl in each well and the wells were covered with a supplied protective coverslip. When the hydrogels had formed in the 3D cell culture experiments, or the cells had started to attach to the surface in the 2D experiments (approximately 1 h), cell culture medium (DMEM supplemented medium for HepG2 and plating medium for hiPS-HEP) was used to wash the hydrogels. The devices were then incubated overnight at 37 °C before three times washing the channels by adding 60 μl cell culture medium (37 °C) from one well and removing the same volume from the opposite well and fixing the devices on a motorized rocker to drive the perfusion back and forth through the device at a frequency of 0.5 Hz. Cell culture medium was changed, and samples were collected every second day by removing 60 μl from each channel followed by an addition of 60 μl cell culture medium to one of the wells and removing the same volume from the opposing well. This procedure was repeated 4 times to replace at least 90% of the cell culture medium, with gentle rocking in between to allow diffusion from the hydrogel to the supernatant. Finally, 60 μl cell culture medium was added to one of the wells on each channel. The experiments were performed in duplicates (4 wells in total for each condition) and samples were stored at −20 °C until analysis.
2.9. Cell morphology, viability and functionality
Cell morphology and cell viability were analysed by incubating the cells in 2 μM calcein-AM, 4 μM ethidium homodimer-1 (Live/Dead viability kit, Molecular Probes), and 5 μg ml−1 Hoechst 33 342 (Molecular Probes) in PBS, for 30 min. Fluorescence images were captured with a confocal microscope (Zeiss LSM700) and processed using ImageJ software (NIH). In addition, the cell functionality was analysed by measuring the hepatocyte secretion of albumin and urea. The supernatants from each condition were pooled and assayed using an ELISA kit (EHALB, Thermo Scientific) and a urea nitrogen colorimetric detection kit (EIABUN, Invitrogen) according to specifications of the suppliers. Samples from a blank experiment with only cell culture medium were subtracted from the urea concentration of HepG2 experiment to account for urea levels in the FBS of the cell culture medium.
3. Results and discussion
3.1. Characterization of HA-PEG hydrogels
The HA-PEG hydrogels are comprised of two components that are crosslinked via a copper-free SPAAC reaction when mixed. The cyclooctyne BCN was grafted to HA to the glucuronic acid carboxyl-moieties using standard carbodiimide chemistry. The final HA-BCN product had a derivatisation degree of 19%, estimated with 1H-NMR (supporting figure S1). The BCN reacts with the azides (N3) on an 8-armed PEG-polymer with terminating N3-moities (p(N3)8), forming a stable 1,2,3-triazole (figure 1(a)). The reaction occurs without any additional compounds under physiological conditions and is fully bioorthogonal resulting in formation of robust hydrogels with tunable viscoelastic properties (figure 1(b)). This hydrogel crosslinking strategy is thus ideal for fabrication of hydrogels for 3D cell culture since cells can be easily encapsulated without compromising their viability. Furthermore, the individual components display low viscosity properties, making them suitable for use in microfluidic devices (supporting figure S5). To evaluate the possibilities to tailor the viscoelastic properties, the ratio of HA-BCN to p(N3)8 was varied and the storage modulus (G') and loss modulus (G'') of the hydrogels were investigated using oscillatory rheology (figure 1(c), supporting figure S8(a)). HA-PEG hydrogels with an excess of N3 moieties did not form hydrogels (not shown) whereas hydrogels with an excess of BCN did, as indicated by G' ≫ G''. The G' increased from about 100 Pa to 1 kPa when increasing the BCN:N3 ratio from 2:1 to 10:1. In addition, the excess of unreacted BCN moieties were available for further modifications with other azide-containing compounds, such as different cell adhesion motifs. To investigate the effect of conjugation of low-molecular compounds to these pendant BCN groups on the mechanical properties of the hydrogels, different concentrations of 2-azidoethanol (EtOH-N3) were added to HA-BCN prior p(N3)8. The resulting viscoelastic properties of the HA-PEG(EtOH-N3) hydrogels were subsequently examined (supporting figures S6, S8(c)). Up to 100 μM EtOH-N3 could be added before any major decrease in G' could be noted. This indicates that it is possible to modify the hydrogel components with other functional molecules without significantly affecting the crosslinking and the mechanical integrity of the hydrogels, which enables flexible introduction of various bio-functionalities. Moreover, since the crosslinking can reduce diffusion of nutrients and waste products though the material as well hamper use of immunostaining techniques, the permeability of the hydrogels to biomolecules was investigated (figure S7). The hydrogels were loaded with either 5(6)-Carboxyfluorescein or Cy5-labelled bovine serum albumin and placed in buffer. The release was monitored over time and indicated a rapid diffusion of the dye labelled molecules in the hydrogels.

Figure 1. (a) Schematic representation of the formation of HA-PEG hydrogels through a strain-promoted alkyne-azide cycloaddition reaction. (b) Photograph of a HA-PEG hydrogel after formation. (c) Frequency sweeps of HA-PEG hydrogels with different BCN to N3 ratios. (d) Gelation time experiment showing the increase of G' over time during gelation of HA-PEG hydrogels (10:1 BCN:N3) at different temperatures.
Download figure:
Standard image High-resolution imageOne desired feature of hydrogels for 3D cell culture applications is the possibility to precisely control the gelation kinetics. The gelation must be rapid enough after addition of cells to prevent cell sedimentation, while still be slow enough to enable proper mixing to ensure homogenous cell distribution throughout the hydrogel [29]. To evaluate the possibility to control the gelation kinetics, 10:1 BCN:N3 HA-PEG hydrogels were formed in situ at different temperatures on the rheometer and the gelation kinetics was monitored using oscillatory rheology at 4 °C, 20 °C and 37 °C (figure 1(d)). At 4 °C, no sol–gel phase transition was observed during a time period of 60 min, as indicated by the absence of increase in G'. In contrast, at 20 °C, an increase of G' to approximately 500 Pa was reached within the same time span although G' continued to increase after 60 min. At 37 °C a more rapid gelation could be seen, reaching approximately 1500 Pa at 60 min. The G' levelled out after 45 min at 37 °C, indicating full gelation. It is thus possible to precisely control the gelation kinetics of the HA-PEG hydrogels by varying the temperature. In addition to enable homogenous incorporation and encapsulation of cells, the possibility to control gelation kinetics also facilitates various biofabrication strategies (e.g. bioprinting) as well as assembly of the hydrogel in microfluidic systems, which is vital for organ-on-a-chip applications.
3.2. Comparison of agarose, alginate and HA-PEG hydrogels
The HA-PEG hydrogels were compared with the well-established 3D cell culture scaffolds agarose and alginate hydrogels. Although more physiological relevant hydrogels do exist, both agarose and alginate consist of a non-sulfonated polysaccharide backbone similar to hyaluronan. Also, both agarose and alginate are commonly used for 3D cell culture of a wide range of cell types, including HepG2 cells [30, 31]. To make sure that the final viscoelastic properties were in the same range in all the 3D cell culture experiments, the viscoelastic properties of hydrogels with different polymer concentrations were examined with oscillatory rheology (figures 2(a)–(c), supporting figure S8(b)). From the rheological assessment, 1% Agarose, 1% Alginate and 2% HA-PEG was chosen for further cell experiments as these were in the same G' range (102–103 Pa). Furthermore, examination of the morphology using SEM showed similar structural features in all hydrogels (figures 2(d)–(f)).

Figure 2. (a)–(c) Frequency sweeps of (a) agarose, (b) alginate and (c) HA-PEG hydrogels at different concentrations. (d)–(f) SEM micrographs of (d) agarose, (e) alginate and (f) HA-PEG hydrogels showing the morphology of each hydrogel.
Download figure:
Standard image High-resolution image3.3. Liver-on-a-chip design and setup
To optimize the performance of the liver-on-a-chip device, the containment and fluidic device properties were adapted to the properties of the hydrogels with encapsulated hepatocytes. This necessitated a design of the chip that met two decisive design criteria, (1) to allow perfusion of cell culture medium over the hydrogel to allow exchange of nutrient media and other analyte components with and from the encapsulated hepatocytes, and (2) allow sampling of the perfused media for analysis without affecting the hepatocytes or the hydrogel. These criteria were met by a commercially available 3D cell culture device (μ-slide III 3D Perfusion, Ibidi), which consists of two consecutive chambers (2 × 30 μl) in three parallel arrays each connected to media reservoirs for perfusate liquid (figure 3(a)). To allow perfusion, the device was mounted on an automatic rocking table reversing the direction of the perfusate over the hydrogel-containing chambers by the exerted gravitation force. At each turn, the source well was raised approximately 5 mm (6° angle) above the receiving well and the flow direction was reversed at a frequency of 0.5 Hz. This setup ensured that most of the cell culture medium was transferred from the source well while keeping air away from the hydrogel chambers, hence meeting the criteria of perfusion through the device. Furthermore, the localization of wells adjacent to the hydrogel chambers allowed sampling of the perfusate without interfering with the hepatocytes or disrupting the hydrogel which made the device fulfil the second criteria.

Figure 3. (a) Schematic representation of the liver-on-a-chip device and setup. The device was put on an automatic rocker table to allow perfusion of media and nutrients during cell culture. (b) Depiction of the HepG2 3D cell culture experiments with HA-PEG hydrogels. The HepG2 cells were added to media-suspended p(N3)8 prior addition of HA-BCN. (c) Depiction of the hiPS-HEP 3D cell culture experiments with HA-PEG hydrogels. The hiPS-HEP cells were added to media-suspended p(N3)8 prior addition of HA-BCN. In experiments using either linRGD or cRGD peptide, the HA-BCN was preincubated with 1 μM of corresponding peptide for 1 h prior adding the HA-BCN(RGD) component to the hiPS-HEP/p(N3)8 mixture.
Download figure:
Standard image High-resolution image3.4. Performance of hydrogel-encapsulated HepG2 cells in the liver-on-a-chip
To investigate the ability for the liver-on-a-chip device to maintain hydrogels with encapsulated hepatocytes with high viability and sustained functionality, the device was first evaluated with hydrogel encapsulated HepG2 cells (figure 3(b)). The morphology, viability and functionality of the HepG2 cells in the HA-PEG hydrogel were compared to agarose and alginate, both known to be compatible with hepatocytes [32–34]. The HepG2 cells formed characteristic spheroids in all hydrogels, typically within 3 d of culture with increasing size over time due to cell proliferation. The viability of the cells was confirmed with a live/dead assay that showed spheroids containing mainly living cells (figure 4(a)). To further confirm that the functionality of the HepG2 cells were maintained, secretion of the hepatocyte characteristic products albumin and urea were analysed after 3, 7 and 9 days of culture. The supernatant of the control 2D culture of cells in the liver-on-a-chip device contained higher levels of both albumin and urea compared to all the 3D cultured cells (figure 4(b)). However, the comparison is uncertain as the growth rate between cells in 2D and 3D might differ significantly [2]. In addition, metabolites and secreted proteins may be trapped in the hydrogel matrix e.g. due to different pore structures of the hydrogels, which thus affects the concentration recorded in the supernatant. Nevertheless, between the hydrogel encapsulated cells, where the same number of cells were seeded, the highest levels of albumin and urea were produced by cells in alginate after 9 and 3 days respectively (table 1). However, detectable levels of both products could be measured in agarose and HA-PEG hydrogels as well. The liver-on-a-chip device could thus maintain the hydrogel encapsulated hepatocytes with high viability and sustained functionality over an extended period of time.

Figure 4. Viability, morphology and functionality of HepG2 cells after 9 days of culture. (a) HepG2 cells formed spheroids of viable cells in all hydrogels as shown by light microscopy (left, 10× objective, scale bars indicate 200 μm) and confocal microscopy (20× objective, scale bars indicate 50 μm) with dyes to show viable cells (green), dead cells (red), and nuclei (blue). (b) Albumin and (c) urea production by HepG2 after 3, 7, and 9 days at different conditions. Supernatants from 4 wells were pooled and analysed in duplicates.
Download figure:
Standard image High-resolution imageTable 1. Summary of the 3D cell culture experiments. The viability and the albumin- and urea production were compared for each cell type and scored based on how they performed in different hydrogels indicated by a relatively high cell viability/production of albumin or urea (++), a moderate cell viability/detectable production of albumin or urea (+), or a low cell viability/no detectable production of albumin or urea (−).
| Hydrogel | ||||||
|---|---|---|---|---|---|---|
| Cell type | Analysis | Alginate | Agarose | HA-PEG | HA-PEG(linRGD) | HA-PEG(cRGD) |
| HepG2 | Morphology | Spheroids | Spheroids | Spheroids | N/A | N/A |
| Live/Dead | ++ | ++ | ++ | N/A | N/A | |
| Albumin | ++ | + | + | N/A | N/A | |
| Urea | ++ | + | + | N/A | N/A | |
| hiPS-HEPs | Morphology | Small aggregates | Small aggregates | Small aggregates | Small aggregates | 3D constructs |
| Live/Dead | − | − | − | − | + | |
| Albumin | − | − | − | − | + | |
| Urea | − | − | − | − | − | |
3.5. Performance of hydrogel-encapsulated hiPS-HEP cells in the liver-on-a-chip
Similar to primary hepatocytes cultured in vitro, induced pluripotent stem cell derived hepatocytes are limited in their proliferation capabilities [35, 36]. A crucial step is therefore to seed the cells in a procedure that maintain a high viability of the cells. Thus, we hypothesised that the HA-PEG hydrogel bioorthogonal crosslinking strategy would be an excellent option for 3D cell encapsulation of hiPS-HEP cells. Moreover, by exploiting the possibility to incorporate cell adhesion motifs using SPAAC, we further hypothesised that the hiPS-HEPs would be able to attach and migrate in the hydrogel, leading to improved cell functionality. To test our hypothesises, 1 μM RGD in either linear (linRGD) or cyclic (cRGD) form were grafted onto HA-BCN prior addition of p(N3)8 and hiPS-HEPs (figure 3(c)). The cyclic form of RGD has previously been shown to have higher affinity for cell integrins, compared to the linear form, due to its more sterically confined conformation [37–40]. The hiPS-HEP cells where cultured in the different hydrogels and on HEP Coat in 2D for 13 d and the morphology and viability were qualitatively examined after 13 d using a live/dead staining. As hypothesized, the confocal micrographs indicate how the survival of hiPS-HEP is dependent on the possibility of the cells to attach to either the 2D surface, the hydrogel or to each other (figure 5). While small aggregates of viable cells could be found sporadically in HA-PEG, HA-PEG(linRGD), alginate, and agarose hydrogels, the hiPS-HEP cells encapsulated in the HA-PEG(cRGD) formed large structures of viable cells that had migrated throughout the hydrogel. Furthermore, analysis of the large cell structures by 3D confocal microscopy showed a distinct difference in the z-orientation of the hiPS-HEP encapsulated and cultured in HA-PEG(cRGD) as compared to cells cultured on HEP Coat in 2D, clearly showing that the hiPS-HEP did grow in true 3D constructs within the HA-PEG(cRGD) hydrogel (figure 6). Moreover, to evaluate the functionality of 3D cell cultured hiPS-HEP cells their albumin and urea excretion were monitored. Albumin expressed and excreted by hiPS-HEPs was only detected in the supernatants of 2D cultures and in the HA-PEG(cRGD) hydrogels (figure 7).
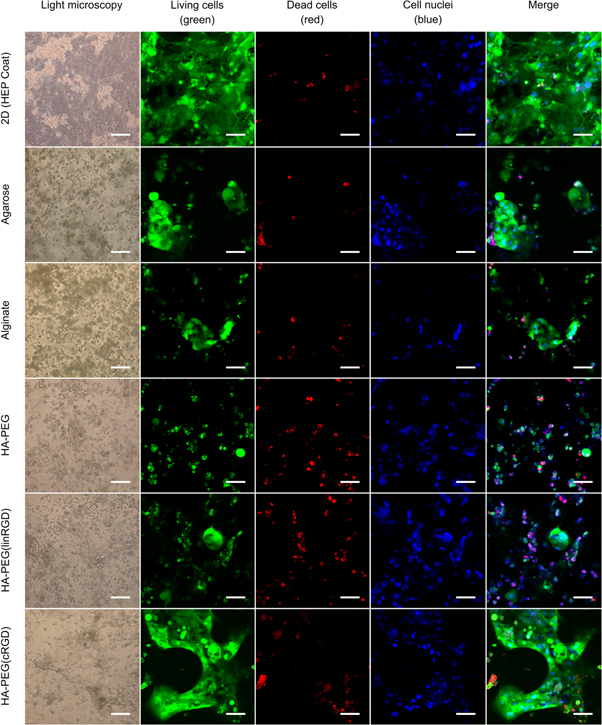
Figure 5. Viability and morphology of hiPS-HEP cells after 13 days of culture. Cell viability seems to be dependent on the cell's ability to attach to the surface, the hydrogel, or to each other. Large structures of hiPS-HEPs were formed in HA-PEG(cRGD). The micrographs were captured after 13 days of culture by light microscopy (left, 10× objective, scale bars indicate 200 μm) or confocal microscopy (20× objective, scale bars indicate 50 μm) with dyes to show viable cells (calcein, green), dead cells (ethidium homodimer-1, red), and nuclei (Hoechst 33 342, blue).
Download figure:
Standard image High-resolution image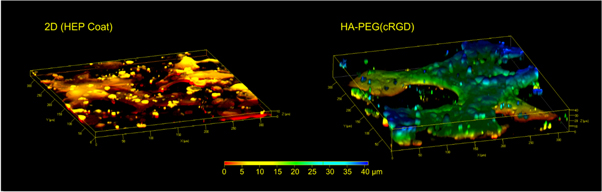
Figure 6. Z-stack of images obtained by confocal microscopy reveals the flat shape of hiPS-HEPS when cultured on a 2D substrate (left) compared to the 3D constructs of the cells in a HA-PEG(cRGD) hydrogel (right). The colours represent the height above the 3D cell culture device chamber bottom, i.e. the Z-axis, from 0 μm (red) to 40 μm (blue).
Download figure:
Standard image High-resolution image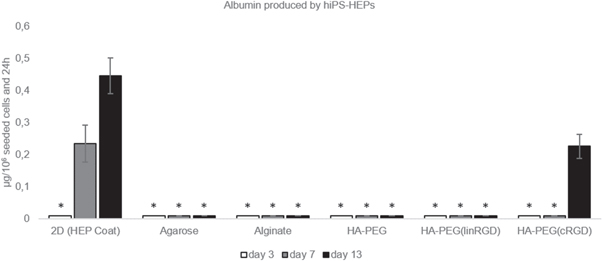
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. Albumin produced by hiPS-HEP on HEP Coat in 2D and in the different hydrogels. * indicates that the albumin concentration levels were too low for reliable quantification. Supernatants from 4 samples were pooled and analysed in duplicates.
Download figure:
Standard image High-resolution image{kind=link}
Albumin was quantified earlier and at higher concentrations when cultured on HEP Coat in 2D as compared to the hydrogels, potentially as a result of slower diffusion of albumin in the hydrogels. Nevertheless, between the different hydrogels, the higher expression of albumin in the HA-PEG(cRGD) hydrogel compared to the other hydrogels correlates with the relatively high viability of the hiPS-HEP cells cultured under these conditions. In addition, traces of urea secreted from hiPS-HEPS were detected in all hydrogels but at levels too low for reliable quantification. Although indications of a higher ratio of viable hiPS-HEPs were found in the HA-PEG(cRGD) hydrogel compared to the other hydrogels, the total number of cells might need to be increased to be able to quantify urea. Another explanation of the low urea and albumin production could be that the combination of cell-binding motifs in the proprietary HEP Coat used for the 2D experiments differs from the pure cRGD cell-binding motif in the hydrogel. Functional behaviour of cells has previously been reported to change depending on the cell-binding motif, or the combination of several different cell-binding motifs, used for the cell culture [41, 42]. Furthermore, as for the HepG2 experiments, the recorded concentration of albumin and urea in the supernatant might be affected by different pore structures of the hydrogels. It should also be noted that cell viability and cell functionality might be enhanced if RGD motifs are grafted in agarose and alginate hydrogels. Based on the results from our experiments, it is advised to use a cyclic form rather than the linear version of the RGD peptide for hiPS-HEPs. However, other types of cell-binding motifs might also be of interest to investigate to obtain an enhanced cell viability and cell functionality in hydrogels. Such experiments would be possible to conduct with the modular approach to form hydrogels presented in this article.
To summarize, the differences in cell response and function in the hydrogels are listed in table 1. The viability and quantifiable production of albumin from hiPS-HEP cultured in HA-PEG(cRGD) confirms our hypothesis that incorporation of well-defined cell adhesion motifs are needed for efficient 3D cell culture of these cells in hydrogels. Moreover, the simplicity and gentleness of the SPAAC crosslinking procedure of the HA-PEG(cRGD) was favourable for the non-proliferating hiPS-HEPs. In contrast, the immortalized HepG2 cells could survive and function in all hydrogels tested in this study. Hence, the choice of cell type should be considered when demonstrating materials such as hydrogels intended to be part of organ-on-a-chip devices.
4. Conclusion
In this paper we have shown the advantages of using a modular hyaluronan-PEG based hydrogel modified with RGD peptides for 3D cultures of hepatocytes in a liver-on-a-chip setup. The hydrogels were fabricated using a bioorthogonal strain-promoted copper-free click-chemistry approach that enabled both tuning of the viscoelastic properties and biofunctionality. The hydrogels were benchmarked with commonly used alginate and agarose hydrogels. Encapsulated HepG2 cells were viable and functional for more than 9 d in the different hydrogels. However, non-proliferating induced pluripotent stem cell line hiPS-HEPs required the presence of cell adhesion motifs in the hydrogel in order to survive and function over a time span of 13 d. Linear and cyclic RGD motifs were grafted to BCN groups in the HA component, where the latter resulted in a pronounced increase in hiPS-HEP proliferation and albumin secretion as compared to non-functionalized hydrogels. Our results show that the hydrogel crosslinking method and possibilities for cell attachment should be carefully considered for stem cell derived cells when choosing 3D cell culture platform. Optimization of viscoelastic properties and concentrations of cell adhesion motifs are still needed to further improve the HA-PEG hydrogel system to meet the exact requirements of the cells. This work is greatly facilitated by the flexible and modular hydrogel fabrication strategy to enable development of 3D cell culture system for sensitive stem cell derived cells. Furthermore, the combination of this hydrogel system with the current device setup will further facilitate design of physiological relevant liver-on-a-chip platforms.
Acknowledgments
This research was funded by the EU Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115439 (StemBANCC project), the resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007-2013) and contribution in kind from EFPIA companies. The authors do also acknowledge the financial support from the Swedish Research Council (VR) (B0431901), the Swedish Foundation for Strategic Research (SFF) (FFL15-0026), the Carl Trygger Foundation (CTS15:79), The Knut and Alice Wallenberg Foundation (Wallenberg Academy Fellow KAW 2016.0231), and the Swedish Government Strategic Research Area in Materials Science on Functional Materials at Linköping University (Faculty Grant SFO-Mat-LiU No. 2009-00971).