
Abstract
Variation in the formation energy of stacking faults (SFs) with the contamination of Na atoms was examined in Si crystals with different Fermi levels. Na atoms agglomerated at SFs under an electronic interaction, reducing the SF formation energy. The energy decreased with the decrease of the Fermi level: it was reduced by more than 10 mJ/m2 in p-type Si, whereas it was barely reduced in n-type Si. Owing to the energy reduction, Na atoms agglomerating at SFs in p-type Si are stable compared with those in n-type Si, and this hypothesis was supported by ab initio calculations.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Photovoltaic (PV) power plants, which are based on sustainable and cost-competitive technologies, are widely recognized as a clean energy source that contributes to reducing global environmental problems involving CO2 emission. They are expected to provide a PV energy of 4.7 TW by 2050,1) which will correspond to a 16% share in the global electricity mix. The reliability of PV technologies, as well as the improvement of the PV efficiency and the reduction of the manufacturing cost, has attracted attention for future-oriented development.
In megawatt-scale PV power plants, numerous PV modules — the majority of which are fabricated with p-type Si solar cells2,3) — are connected in series to generate a high voltage and power. High-voltage stresses up to several hundreds of volts, are inevitably applied between each cell and its module frame. As reported by Luo et al.,4) these stresses can cause a catastrophic power drop in the module (called a potential-induced degradation, PID).5) The PID mechanism in p-type Si solar cells has been intensively studied, revealing that the key factor of the PID is Na atoms existing inside6–9) and on10) the solar module encapsulation (such as the cover glass and SiNx antireflection coating layer), as well as on Si cells.11) During operation, these Na atoms accumulate at the SiNx/Si interface and agglomerate at stacking faults (SFs) in Si cells expanding from the interface.6) Na atoms are preferentially located on SFs,12,13) creating a conductive layer.12) This can provide a high conductivity between the n-doped emitter and the p-doped base when the SF penetrates the n–p junction, resulting in a reduction of the shunt resistance, which degrades the solar-cell performance.6) Indeed, the PID power loss correlates with the amount of Na atoms.14) Owing to the PID stress under the influence of Na penetration, these SFs nucleate from microscopic defects at the interface,15) such as dislocations,16) and grow via the diffusion of Na atoms in the SFs decorated with Na atoms,12) which is significantly faster than that in bulk Si.17) The Na penetration processes are modified depending on the doping level in Si cells.18) Additionally, the amount of Na atoms agglomerating at SFs in p-type Si appears to be larger than that for n-type Si.19) These results suggest that the formation energy of SFs depends on the Fermi level, as well as on the existence of Na atoms. In this work, we examined the interaction of Na atoms with SFs in Si crystals having different Fermi levels, leading to a reduction in the SF formation energy. The electronic interaction of Na atoms with SFs depending on the Fermi level was determined via transmission electron microscopy (TEM) and first-principles calculations based on density functional theory. Our microscopic findings provide guidance for designing PID-resistant Si solar cells.
The specimens were dislocation-free Si single crystals grown via the Czochralski method: p-type Si (doped with B atoms with a carrier concentration of c = 8 × 1018 cm−3), n-type Si (doped with P atoms with c = 3 × 1019 cm−3), and nominally undoped n-type Si (c = 3 × 1013 cm−3).20) SF ribbons bound by pairs of partial dislocations (density of approximately 109 cm−2) were intentionally introduced into the crystals by applying a compressive stress in an Ar gas atmosphere at an elevated temperature of 1,173 K for 0.5 h.20) Each crystal and a Na sample (Nippon Soda, purity; 99.95%) were sealed within a stainless-steel tube containing Ar gas, and the crystal was contaminated with Na atoms by heating the tube in a furnace at 973 K for 5 h.21) The width of each SF ribbon wSF was estimated via dark-field (DF) TEM,22) as a function of the line orientation α defined as the angle of b to u, where b is the sum of the Burgers vectors of the dislocations bounding the SF ribbon, and u is a vector parallel to the dislocations. The apparent energy for the formation of SFs ESF was then calculated with wSF(α), by using anisotropic elasticity theory considering the deviation parameter.23)
The stability of Na defects at different Fermi levels was examined via density functional theory calculations. We constructed a supercell with an SF consisting of 224 Si atoms, whose cell volume was 1.33 × 4.40 × 0.77 nm3, and introduced 1 Na interstitial defect to the supercell. In addition to neutral defects [Na(0)], positively charged defects [Na(+)] and negatively charged ones [Na(−)] were considered by either subtracting or adding one electron. We used the projector augmented-wave method implemented in the VASP code.24–26) Plane waves with an energy cutoff of 500 eV were used to expand the wave functions. The exchange–correlation energy was calculated using a revised version of the generalized gradient approximation of Perdew–Burke–Ernzerhof (GGA-PBEsol).27) A Γ-centered k-point mesh of 1 × 2 × 4 was used for integration of the Brillouin zone. The electrons of 3s23p2 for Si and 3s1 for Na were treated as valence electrons. The atomic positions were optimized until the force on each atom was <0.01 eV/A.
Before the contamination with Na impurities, wSF(α) distributed in the range of 4 to 8 nm; it was maximized for α ≈ 90° and minimized for α ≈ 0°, owing to the balance between the SF energy and the elastic energy due to partial dislocations.23) The average of wSF(α) at α increased in Si crystals contaminated with Na atoms. Figures 1(a) to 1(b) show the typical variation observed in p-type Si crystals. The ratio of wSF(α) before contamination to that after Na contamination was almost independent of α [Fig. 1(c)]. Accordingly, as shown in Fig. 1(d), the ESF estimated with wSF(α) decreased by a constant rate regardless of α.

Fig. 1. DF-TEM images of an SF with α ≈ 90° in p-type Si viewed along the SF normal (with 200-keV electrons having g = 220 under the g/4g condition) (a) before and (b) after contamination with Na atoms. (c) SF width wSF(α) and (d) SF formation energy ESF before (triangles) and after (circles) Na contamination.
Download figure:
Standard image High-resolution imageAn SF ribbon can widen because of the reduction of the electronic energy due to the agglomeration of impurities at the ribbon.20) Additionally, impurity atoms can agglomerate at the pair of partial dislocations binding the SF ribbon, reducing the electronic and elastic energies, and this energy reduction causes the variation of wSF(α). However, although the ability of impurity agglomeration at dislocations, which is related to the electronic structure and the elastic strains around the dislocation core, depends on α, Fig. 1(d) shows that ESF is independent of α. Thus, the effects of Na agglomeration at partial dislocations are negligible in the present experiments. Additionally, the Coulomb repulsion in a pair of partial dislocations,28) charged negatively via the displacement of the Fermi level by Na donors (Patel effect),29) would be ignored in p-type Si crystals heavily doped with B atoms. These results indicate that Na atoms agglomerate at SFs owing to an electronic interaction, and this interaction results in the reduction of ESF.
Figure 2 shows the energy reduction in ESF induced by Na contamination in Si with different Fermi levels. The reduction in p-type Si was >10 mJ/m2: the average ESF of 59 mJ/m2 before Na contamination decreased to 47 mJ/m2 [Fig. 2(a)]. Similarly, the ESF for undoped Si was reduced by Na contamination, even though the energy reduction was a few mJ/m2 [Fig. 2(b)]. Meanwhile, ESF was barely reduced in n-type Si30) [Fig. 2(c)]. These results suggest that the reduction in ESF due to Na contamination increases with the decreasing Fermi level. That is, Na atoms agglomerating at SFs in p-type Si are stable compared with those in n-type Si, because of the electronic interaction of Na atoms with SFs.

Download figure:
Standard image High-resolution image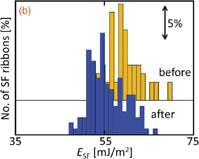
Download figure:
Standard image High-resolution image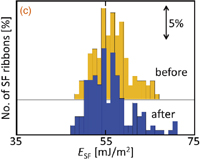
Fig. 2. Estimated SF energy ESF before and after Na contamination in (a) p-type, (b) undoped, and (c) n-type Si crystals.
Download figure:
Standard image High-resolution imageThe aforementioned hypothesis was supported by ab initio calculations. Because no Na precipitate was observed at SFs via TEM, we assumed that isolated Na atoms agglomerate at SFs. We did not consider Na atoms at substitutional sites, because their formation energy would be rather high.33) We confirmed that Na atoms are most stable at the "open sites in SFs" indicated by the square in Fig. 3, and they are located at the tetrahedral sites in Si crystals free from SFs, as previously reported.12) The binding energy of Na atoms to SFs, Ebind, is determined as the difference between the total energy of the supercell in which the Na atom is located at an "open site in SFs" and that at a tetrahedral site far from the SF, as indicated in Fig. 3. The binding energy was estimated to be 0.7 eV regardless of the charge state, indicating that the binding energy is independent of the Fermi level. The formation energy of Na defects at the tetrahedral sites free from SFs, ETD, depends on the Fermi level: the energy in p-type Si is lower than that in undoped and n-type Si.33) The formation energy of Na defects at the "open sites in SFs", Eopen, can be approximated as ETD − Ebind; therefore, the energy in p-type Si is lower than that in undoped and n-type Si. Na atoms with a low ionization energy exist as Na(+) in p-type Si by capturing holes, as theoretically predicted,33) resulting in a low Eopen for p-type Si. That is, the interaction of ionized Na atoms with SFs results in a large reduction of the SF energy in p-type Si.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fig. 3. Supercell of a SF consisting of Si atoms (indicated by circles). The atomic sites for interstitial Na indicated by the square and the triangle are the "open site in SFs" and the tetrahedral site, respectively, used for our calculations.
Download figure:
Standard image High-resolution image{kind=link}
Here we discuss the agglomeration process of Na atoms at SFs. It is hypothesized that SFs are formed at the SiNx/Si interface via the agglomeration of Na atoms and expand towards the n–p junction under PID stresses.15) We clarified experimentally that the SF formation energy is reduced in Si contaminated with Na atoms, as theoretically predicted.12,13) Once contaminated SFs are formed, Na atoms can diffuse preferentially along the SFs because the activation energy for Na diffusion in the SFs is lower than that in bulk Si.12) Under a constant PID stress, the expansion rate of SFs determined by the Na diffusion increases with the increasing distance from the SiNx/Si interface, owing to the reduction in the SF formation energy depending on the Fermi level (Fig. 2). In particular, it is expected that the expansion rate of SFs increases rapidly after the SFs penetrate the n–p junction. Such a rapid increase in the expansion rate may result in an abrupt increase in the power loss in solar cells, as previously reported.5,14,16,34) Indeed, it is speculated that Na diffusion into the n–p junction induces irreversible PID processes.34) Meanwhile, the expansion rate decreases with the concentration of n-type dopants, which may be related to the suppression of the PID in Si heavily doped with P atoms18) and that in Si cells under photo-illumination.35) It is also expected that the amount of Na atoms agglomerating at SFs in p-type Si is larger than that in n-type Si, as observed in a solar cell after PID.19) Our results suggest that the PID progression in p-type Si solar cells depends on the distribution of dopant atoms around the n–p junction, as well as on external factors such as the applied voltage and temperature.4)
As reported by Luo et al.,4) the PID in p-type Si solar cells can be recovered by annealing and/or by applying a reverse potential, and the recovery process depends on the PID history of the cells. It is hypothesized that the process is controlled by the out-diffusion of Na atoms from SFs.8) Because of the out-diffusion, SFs decorated with Na atoms are converted into undecorated SFs, which are electrically inactive.8) Some p-type Si solar cells exhibit incomplete recovery, probably due to residual Na atoms in SFs.34,36) A SF decorated with Na atoms is observed in a recovered solar cell.37) These complicated behaviors of Na atoms may correlate with the formation energy of the SFs decorated with Na atoms, which depends on the Fermi level (Fig. 2). The energy near the SiNx/Si interface, where the concentration of n-type dopants is high, is comparable to the formation energy of SFs without Na atoms. That is, the interaction of Na atoms with SFs is weak. This weak interaction assists the out-diffusion of Na atoms near the interface. With an increasing distance from the interface, the SF formation energy decreases, and the stability of Na atoms at SFs increases. This stability disturbs the out-diffusion of Na atoms in SFs, especially around the n–p junction. Further examination of the spatial distribution of dopants and Na atoms is necessary for confirming the model.
In conclusion, the formation energy of SFs decorated with Na atoms was examined in Si crystals with different Fermi levels. The estimated energy decreased with the decrease of the Fermi level: the energy in p-type Si was reduced by >10 mJ/m2, and the energy in n-type Si was comparable to the formation energy of SFs without Na atoms. These results suggest that the PID progression and recovery in p-type Si solar cells, which is caused by the Na motion in SFs, correlates with the distribution of dopant atoms around the n–p junction.
Acknowledgments
A part of this work was supported by JSPS KAKENHI for Scientific Research (B) Grant Number JP15H03535 (2015–2018) and JST CREST Grant No. JPMJCR17J1 (2017–2023). The TEM analyses were partially supported by the Laboratory of Alpha-Ray Emitters in the Institute for Materials Research (IMR), Tohoku University, under the Inter-University Cooperative Research Program in IMR.