

Abstract
New sulforhodamine-based fluorescent 'turn-on' probes have been developed for the direct imaging of cellular hypoxia. Rapid access to this novel class of water-soluble 'azobenzene-caged' fluorophores was made possible through an easily-implementable azo-coupling reaction between a fluorescent primary arylamine derived from a sulforhodamine 101 scaffold (named SR101-NaphtNH2) and a tertiary aniline whose N-substituents are neutral, cationic, or zwitterionic. The detection mechanism is based on the bioreductive cleavage of the azo bond that restores strong far-red fluorescence (emission maximum at 625 nm) by regenerating the original sulforhodamine SR101-NaphtNH2. This valuable fluorogenic response was obtained for the three 'smart' probes studied in this work, as shown by an in vitro assay using rat liver microsomes placed under aerobic and then under hypoxic conditions. Most importantly, the probe namely SR101-NaphtNH2-Hyp-diMe was successfully applied for imaging the hypoxic status of tumor cells (A549 cells).
Export citation and abstract BibTeX RIS
1. Introduction
Rhodamine dyes and related photoactive compounds (i.e. rhodols and rosamines) are wide-spread fluorophores [1]. They are currently used as reporters in various biological and bioanalytical applications that involve biosensing [2] and biolabeling operations [3]. This widespread use is due to the fact that such xanthene-based fluorophores combine numerous attractive and valuable properties including: (1) High brightness both in organic and aqueous media often associated with a marked photostability, (2) very high chemical stability, especially under harsh conditions of pH and temperature, and (3) easy modulation of the fluorescence properties through the spirocyclic/open-ring switching mechanism or through the reversible chemical modification (mainly, amidation) of the aniline moieties [2]. Thus, a huge number of 'turn-on' reactive probes (also known as fluorescent chemodosimeters) based on a rhodamine (or rhodol/rosamine) scaffold have been developed for the detection and/or imaging of various (bio)analytes including enzymes, biomolecules, polluting chemicals, cations, and anions [4, 5]. Current research efforts are primarily devoted to the design of far-red to near-infrared (NIR) emitting rhodamines [6, 7] with significant water solubility, cell permeability, and bioconjugation ability to facilitate the practical implementation of these fluorogenic xanthene-based probes in living cells and organisms and to improve their bioimaging performances [8]. In this context, we recently explored the chemistry and fluorescent biosensing applications of a novel class of sulfoxanthene dyes, which can be regarded formally as unsymmetrical derivatives of sulforhodamine 101 (SR101, also known as Texas Red for its sulfonyl chloride derivative) [9, 10]. Among the various examples of the SR101 analogs synthesized, two of them, bearing a primary aniline and named SR101-110 and SR101-NaphtNH2, are particularly interesting because they can be readily converted into latent fluorophores (also known as pro-fluorophores)5 [11] through a single chemical transformation of their NH2 group into a (bio)analyte-reactive quenching moiety (typically, amidation or conversion into 'self-immolative' carbamate). Interestingly, the azo chemistry was also considered for the reversible masking of primary anilines and it was shown that the direct conjugation of -N = N- group to the conjugated system of a rhodamine/rhodol fluorophore is an effective way of switching off its fluorescence (figure 1 for the structures of the corresponding azo-based fluorescent probes) [12, 13]. This quenching is ascribed to two major processes: (1) The locking of the xanthene platform in a non-conjugated spironolactone form and (2) the ultrafast E/Z isomerization of the -N = N- bond after photoexcitation [14]. The green fluorescence of azo-based probes 1 and 2 is readily restored upon bioreduction (mediated by reductases in bacteria) and reaction with endogenous sulfides (gasotransmitter hydrogen sulfide, H2S), respectively. Further extension of this fluorogenic reduction process to other fluorophore scaffolds, namely 2Me rhodamine green (2Me RG) and its silicon analog 2Me SiR600, was explored by Piao et al and promising hypoxia imaging agents, namely MAR and MASR, were obtained (figure 1) [15]. Hypoxia is a pathological condition in which tissues lack the oxygen required for cells for normal metabolization processes [16]. In some solid tumors, for example, the median oxygen concentration has been reported to be around 4%, and locally it may even decrease to 0% [17]. In oncology, tumor hypoxia is often associated with poor response to treatment protocols and poor clinical outcome. This strongly supports the need to identify tumors with high hypoxic fractions to implement hypoxia-directed treatments and to optimize tumor treatments that are oxygen-sensitive (i.e. photodynamic and radiotherapy). Therefore, it is of considerable clinical significance to develop contrast agents for imaging the hypoxic status of tumor cells [18]. It is known that hypoxia can induce accelerated bioreductive reactions and result in greater expression of intracellular reductases, such as azoreductase (AzoR), cytochrome P450 reductase, quinone reductase (e.g. DT-diaphorase), and nitroreductase (NTR). Consequently, one of the preferred approaches for the detection and localization of hypoxia is based on the use of 'turn-on' reactive probes selectively activated by one of these redox enzyme classes [19–22]. Despite the large number of reductase-activatable fluorescent probes published during the last few years and which are often based on the NTR-mediated reduction of a nitro-aryl moiety acting as a reactive quenching unit [22], less attention has been paid to the optimization of their photophysical (i.e. fluorescence brightness under physiological conditions and in the far-red or NIR range, photo(chemical) stability) and physicochemical (e.g. water solubility, cell permeability, etc) properties. Thus, we thought it worthwhile to explore novel reductase-sensitive fluorescent probes based on the far-red emitting sulforhodamine dye SR101-NaphtNH2, by readily converting its primary aniline to azobenzene moiety, whose -N = N- bridge cleavage will act as a fluorescence trigger in response to AzoR in hypoxia environments (figure 1). In addition to the good spectral features of the sulfoxanthene scaffold in physiological conditions, factors such as water solubility and the net electric charge of the probe can be easily tuned through 'post-synthetic' derivatization of the 'convertible' azo-fused 4-(N,N-dialkylamino)phenyl group (e.g. di-azido derivative modified through copper-catalyzed azide-alkyne cycloaddition (CuAAC) reaction, also known as 'click reaction') [23] thus obviating time-consuming de novo synthesis. In this paper, we report the straightforward chemical synthesis and photophysical characterization of three members of this novel class of 'turn-on' reactive far-red fluorescent probes. In view of their valuable properties such as high specificity, 'OFF–ON' fluorescence response, appreciable solubility, rapid response time, and good biocompatibility, these azo-based probes were next applied for imaging the hypoxic status in A549 tumor cells.

Figure 1. (Top) structures of fluorescent anilines suitable for designing latent fluorophores, recently reported in the literature; (middle) azo-based fluorescent probes recently developed for the biosensing/bioimaging of reductases and gasotransmitter H2S; (bottom) hypoxia imaging agents studied in this work (structures and proposed detection mechanism).
Download figure:
Standard image High-resolution image2. Results and discussion
2.1. Synthesis of azobenzene-caged sulforhodamine dyes
The synthetic strategy currently used to prepare azo dyes relies on the coupling reaction between a diazonium compound and an electron-rich tertiary aniline. The use of a bench-stable, commercially available nitrosating agent, nitrosonium tetrafluoroborate (NOBF4), in a polar aprotic solvent has recently emerged as valuable conditions to perform this SEAr reaction under mild conditions, fully compatible with the moderate stability of a wide range of functionalized anilines [12, 24]. Thus, this diazo coupling methodology appears to be well suited to the conversion of a fluorescent aniline such as SR101-NaphtNH2 into the corresponding azo-based fluorogenic probe. First, and as shown in scheme 1, SR101-NaphtNH2 was readily obtained in a satisfying yield through a concise synthetic route based on the sequential condensation of two different meta-aminophenols (i.e. 8-hydroxyjulolidine and 6-aminonaphthol) with 4-formylbenzene-1,3-disulfonic acid in methanesulfonic acid heated at 150 °C [9]. After isolation by RP-HPLC purification and freeze-drying, TFA salt of SR101-NaphtNH2 was reacted with NOBF4 in dry CH3CN and in the presence of 1 equiv. of TEA (aimed at deprotonating its primary aniline). The resulting diazonium salt (and/or N-nitroso intermediate) was condensed with N,N-dimethylaniline in a mixture of CH3CN and sodium acetate buffer (NaOAc, 0.1 M, pH 4.0) to give the azobenzene-caged sulforhodamine dye SR101-NaphtNH2-Hyp-diMe in a satisfying 51% isolated yield after purification by semi-preparative RP-HPLC (recovered as TEA salt). Its structure was unambiguously confirmed by detailed measurements including NMR and ESI-LRMS analysis (see the experimental section and supporting information). To rapidly obtain azo-sulforhodamine hybrids with enhanced water solubility best suited to biochemical validations with isolated redox enzymes or in living cells, we next planned to synthesize N,N-bis(azidoethyl)diazo pro-fluorophore SR101-NaphtNH2-Hyp-diN3 which can be easily derivatized by 'click chemistry' aimed at imparting the polarity of this azo-based fluorogenic probe (scheme 2). Indeed, previous works from our lab have shown the effectiveness of such a 'post-synthetic derivatization' approach to increase the aqueous solubility of numerous hydrophobic chromophore systems and fluorescent cores by means of simple, efficient, and high-yielding reactions (e.g. Schotten–Baumann amidation, Sonogashira cross-coupling, and CuAAC reactions) involving negatively or positively-charged hydrophilic linkers [25]. In the present case, we have chosen to introduce two positively-charged trialkylmethylammonium groups or two zwitterionic sulfobetaine linkers, whose ionization state is not pH-dependant. Indeed, such polar groups should not affect or, even better, may promote the cell permeability of the resulting water-soluble derivatives [26], which is a key parameter for fluorogenic probes aiming at targeting cellular reductases. The azo coupling between SR101-NaphtNH2 and N,N-di(2-azidoethyl)aniline 3 was performed under the same conditions as those optimized for the synthesis of SR101-NaphtNH2-Hyp-diMe (vide supra). The resulting 'click-convertible' azo dye was readily obtained in a moderate 44% isolated yield and in a pure form by semi-preparative RP-HPLC purification (recovered as TEA salt). The practical implementation of the 'post-synthetic' water-solubilization procedure involved the use of the known terminal alkynes 4 and 5 and standard CuAAC reaction conditions (CuSO4 and sodium ascorbate as the catalytic system) known for not reducing the -N = N- bridge of azo dyes. A mixture of DMF/H2O (1: 1, v/v) was used as the solvent and complete conversion into the 'clicked' azobenzene-caged sulforhodamines SR101-NaphtNH2-Hyp-sulfobetaine and SR101-NaphtNH2-Hyp-ammonium was observed within 2 h. Purification was achieved by semi-preparative RP-HPLC and these water-soluble analogs were recovered as TEA and TFA salt, respectively (isolated yield: 65% and 71% respectively). The spectroscopic data are in agreement with the structures assigned (see the experimental section and supporting information). Sulfonate moieties onto the meso-phenyl ring and possibly polar aniline substituents (N-methylpyrrolidinium and sulfobetaine) make these azo-sulforhodamine hybrids significantly soluble in water and related aqueous buffers, over a concentration range (from μM to mM) fully compatible with their use in bioimaging.
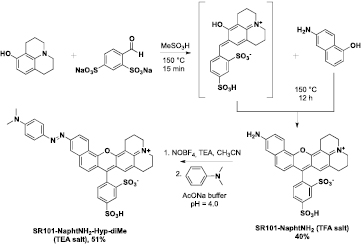
Scheme 1. Synthesis of unsymmetrical sulforhodamine SR101-NaphtNH2 and its conversion into 'azobenzene-caged' derivative SR101-NaphtNH2-Hyp-diMe.
Download figure:
Standard image High-resolution image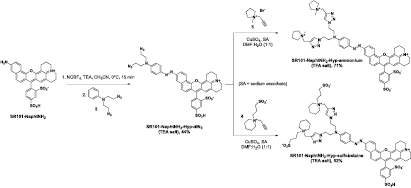
Scheme 2. Synthesis of 'click-convertible' azo dye SR101-NaphtNH2-Hyp-diN3 and its conversion into hydrophilic derivatives SR101-NaphtNH2-Hyp-sulfobetaine and SR101-NaphtNH2-Hyp-ammonium.
Download figure:
Standard image High-resolution image2.2. Photophysical properties of azobenzene-caged sulforhodamine dyes and their response to azoreductase activity
The UV–vis absorption spectra of the three azo-sulforhodamine dyes and parent aniline SR101-NaphtNH2 were recorded in PBS (0.1 M, pH 7.4, simulated physiological conditions), as shown in figure 2. As usually observed for azo-fluorophore hybrids, SR101-NaphtNH2-Hyp-X exhibited a broad absorption spectrum which covers the full visible spectrum, and was characterized by a maximum centered at 570–578 nm according to the aniline substitution pattern. It is interesting to note that the absorption spectra of the more hydrophilic derivatives SR101-NaphtNH2-Hyp-sulfobetaine and SR101-NaphtNH2-Hyp-ammonium are less broad and display a well-defined vibronic structure, which may reflect the beneficial effects of grafted cationic and zwitterionic solubilizing moieties to partly disrupt aggregates in aqueous media. As expected, these three azo-based probes, without exception, were not fluorescent under simulated physiological conditions (ΦF < 0.001) and their emission spectrum exhibits a zero-baseline signal that remains constant over time (see supporting information).
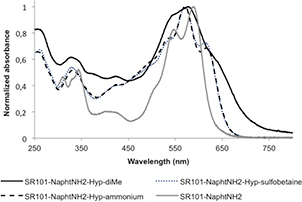
Figure 2. Normalized absorption spectra of unsymmetrical sulforhodamine SR101-NaphtNH2 and its 'azobenzene-caged' derivatives SR101-NaphtNH2-Hyp-X recorded in PBS at 25 °C.
Download figure:
Standard image High-resolution imageTo examine whether the three pro-fluorophores SR101-NaphtNH2-Hyp-X (X = diMe, sulfobetaine, and ammonium) can detect hypoxia in biological systems, we first conducted in vitro fluorogenic assays using rat liver microsomes, which are known to contain a wide range of redox enzymes. After adding NADPH (50 μM) as a cofactor for the reductases to aqueous solutions of SR101-NaphtNH2-Hyp-X (1 μM) in the presence of rat liver microsomes (226 μg/3 mL), a rapid and dramatic increase in red fluorescence intensity at 626 nm (ex. 589 nm) fully consistent with the release of free sulforhodamine SR101-NaphtNH2 was observed only under hypoxic conditions (see figure 3 for the time-course curves and supporting information for the emission spectra recorded after bioreductive activation). Interestingly, the reaction kinetics of SR101-NaphtNH2-Hyp-X are quite similar and little influenced by the grafted cationic or zwitterionic water-solubilizing substituents even if the fluorescence level reached within 1 h for SR101-NaphtNH2-Hyp-ammonium is somewhat lower than that obtained for di-methyl and bis-sulfobetaine derivatives. These preliminary results unambiguously confirm the detection mechanism proposed in figure 1.
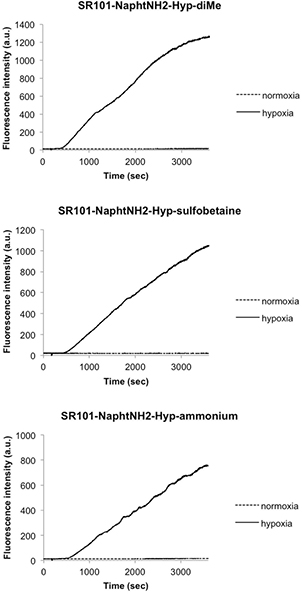
Figure 3. Time-dependent changes in the red fluorescence intensity of SR101-NaphtNH2-Hyp-X (X = diMe, sulfobetaine and ammonium), concentration: 1 μM, in the presence of rat liver microsomes (226 μg/3 mL) under hypoxia or normaxia. Measurements were performed in a potassium phosphate buffer (0.1 M, pH 7.4) containing DMSO (0.1%) as a cosolvent. As a cofactor for reductases, NADPH (50 μM) was added after 3 min. The hypoxic conditions were prepared by bubbling argon gas into the reaction solution for 30 min. The excitation and emission wavelengths were 589 and 626 nm.
Download figure:
Standard image High-resolution image2.3. Fluorescence imaging of hypoxia in A549 cells by azobenzene-caged sulforhodamine dyes
We next applied these probes to living cells to monitor cellular hypoxia. A549 cells (human epithelial lung carcinoma cells) were selected because they are known to express reductases, especially in hypoxic conditions. In our experiments, A549 cells were cultured and subsequently incubated with probes SR101-NaphtNH2-Hyp-X at 37 °C under normoxic (20% O2) and hypoxic (1% and 0.1% O2) conditions for 6 h. Only the cells treated with probe SR101-NaphtNH2-Hyp-diMe under severe hypoxic conditions (0.1% O2) produced a striking bright-red fluorescence (figure 4). Bright spots were observed probably at lysosomes inside the cells. Our assumption put forward to explain the unexpected lack of reductive activation for SR101-NaphtNH2-Hyp-sulfobetaine and SR101-NaphtNH2-Hyp-ammonium is their too large size and/or too high hydrophilic character that limits their cell penetration, required for a close encounter with intracellular produced azoreductases. Finally, the in cellulo activation of SR101-NaphtNH2-Hyp-diMe was compared to those of azo-rhodamines MAR (based on the green-emitting fluorophore 2Me RG) and MASR (based on the red-emitting fluorophore 2Me SiR600), promising hypoxia imaging agents recently reported by Piao et al [15]. A similar fluorescence enhancement inside cells under severe hypoxia (0.1% O2) was obtained for SR101-NaphtNH2-Hyp-diMe compared to MASR, which also exhibits emission in the same spectral range. Conversely, less impressive results than those gained with the green-emitting pro-fluorophore MAR were observed because the fluorescence intensity of the latter increased at oxygen concentrations of 1% or less (figure 5). At this stage, this oxygen concentration dependency of the fluorescence intensity increase of SR101-NaphtNH2-Hyp-diMe and MAR was not easily interpreted.

Figure 4. Fluorescence confocal microscopy images of SR101-NaphtNH2-Hyp-X in live A549 cells (left: Magnified images, right: The differential interference contrast (DIC) images of the corresponding samples are also shown = gray images). The A549 cells were incubated with SR101-NaphtNH2-Hyp-X (X = diMe, sulfobetaine or ammonium) (1 μM) containing DMSO (0.1%) as a cosolvent at various oxygen concentrations (0.1%, 1% and 20%) for 6 h. Scale bar: 30 μm. The excitation and emission wavelengths were 590 and 600–670 nm.
Download figure:
Standard image High-resolution image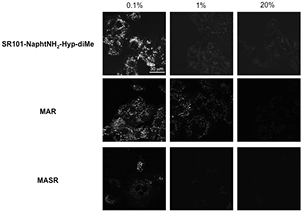
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. Fluorescence confocal microscopy images of SR101-NaphtNH2-Hyp-diMe, MAR or MASR in live A549 cells. The A549 cells were incubated with SR101-NaphtNH2-Hyp-diMe, MAR or MASR (1 μM) containing DMSO (0.1%) as a cosolvent at various oxygen concentrations (0.1%, 1% and 20%) for 6 h. Scale bar: 30 μm. The excitation wavelength and emission range of detection were 488 and 515–553 nm for MAR, 590 and 600–670 nm for SR101-NaphtNH2-Hyp-diMe, and 600 and 620–700 nm for MASR, respectively.
Download figure:
Standard image High-resolution image{kind=link}
3. Conclusions
In summary, we have presented the first practical application of a novel far-red emitting aniline-based fluorophore (SR101-NaphtNH2) in biosensing/bioimaging of hypoxia and azoreductases. This unsymmetrical sulforhodamine dye is readily accessed synthetically and its fluorescence is completely quenched by the conversion of its primary amino group into an arylazo moiety. Interestingly, depending on the nature of the aniline N-substituents (i.e. 2-azidoethyl instead of methyl), it was possible to 'post-synthetically' modify the azo-sulforhodamine scaffold by the CuAAC reaction with hydrophilic terminal alkynes, to readily introduce the N-methylpyrrolidinium or sulfobetaine groups and enhance the water solubility of the resulting azo-based probes. These latter fluorogenic compounds were found to be readily activated by reductases and NADPH under physiological conditions to give back SR101-NaphtNH2, leading to 94-, 45-, and 58-fold fluorescence emission enhancement at ca. 625 nm within 1 h for diMe, sulfobetaine, and ammonium, respectively. The applicability of SR101-NaphtNH2-Hyp-diMe has been demonstrated by visualizing the hypoxic status in A549 cells. The two main advantages of this 'smart' probe compared to the published hypoxia imaging agent MASR, which also exhibits a fluorescence 'OFF–ON' response in the far-red spectral range, are: (1) Ease of synthesis (two steps against four steps from the starting material 6-aminonaphthol and 3-bromo-N,N-dimethylaniline, respectively) [27] and (2) the opportunity to tune its physicochemical properties without resorting to multi-step de novo synthesis. This study confirms the potential use of SR101-NaphtNH2 as a valuable far-red emitting fluorogenic dye in the rapid construction of 'turn-on' reactive probes for chemoselective bioimaging [5].
4. Experimental section
4.1. General
All the chemicals were used as received from commercial sources without further purification unless otherwise stated. Acetonitrile (CH3CN) and triethylamine (TEA) were dried by distillation over CaH2 and over KOH (followed by storage over BaO), respectively. The HPLC-gradient grade CH3CN was obtained from VWR. The phosphate buffer (PB, 100 mM phosphate, pH 7.4), phosphate buffered saline (PBS, 100 mM phosphate + 150 mM NaCl, pH 7.5) and aq. mobile-phases for HPLC were prepared using water purified with a Milli-Q system (purified to 18.2 MΩ cm). The triethylammonium acetate (TEAA, 2.0 M) and triethylammonium bicarbonate (TEAB, 1.0 M) buffers were prepared from distilled TEA and glacial acetic acid or CO2 gas. TFA salt of the unsymmetrical sulforhodamine dye SR101-NaphtNH2, N,N-di(2-ethylazido)aniline 3 and hydrophilic terminal alkynes 4 and 5 were synthesized according to the protocols recently reported by us [9, 12, 28].
4.2. Instruments and methods
1H- and 13C-NMR spectra were recorded with either a Bruker DPX 300 spectrometer or a Bruker Avance III 500 spectrometer. Chemical shifts are expressed in parts per million (ppm) from the residual non-deuterated solvent signal [29]. J values are expressed in Hz. Infrared (IR) spectra were recorded with a universal ATR sampling accessory either on a Perkin Elmer FT-IR Spectrum 100 spectrometer or Bruker Alpha FT-IR spectrometer. Analytical HPLC was performed on a Thermo Scientific Surveyor Plus instrument equipped with a PDA detector. Semi-preparative HPLC was performed on a Thermo Scientific SPECTRASYSTEM liquid chromatography system (P4000) equipped with a UV-visible 2000 detector. Ion-exchange chromatography (for desalting azo-sulforhodamine dyes purified with TEAB as the aq. mobile phase) was performed with an Econo-Pac™ disposable chromatography column (Bio-Rad, #732–1010) filled with an aq. solution of Dowex® 50WX8-400 (Alfa Aesar, ca. 5 g for 15 mg of dye, 15 × 50 mm bed), regenerated using aq. 10% HCl solution and equilibrated with deionized water. Low-resolution mass spectra (LRMS) were obtained with a Finnigan LCQ Advantage MAX (ion trap) apparatus equipped with an electrospray (ESI) source. UV-visible spectra were obtained on a Varian Cary 50 scan spectrophotometer by using either a rectangular quartz cell (Varian, standard cell, Open Top, 10 × 10 mm, 3.5 mL) or a quartz micro cell (Hellma, 108.002-QS, light path: 10 mm, 500 μL). Fluorescence spectroscopic studies (emission/excitation spectra) were performed with a Varian Cary Eclipse spectrophotometer with a semi-micro quartz fluorescence cell (Hellma, 104F-QS, 10 × 4 mm, 1400 μL). Emission spectra were recorded under the same conditions after excitation at the corresponding wavelength (excitation and emission filters: Auto, excitation and emission slit = 5 nm). In figure 3, the fluorescence spectroscopic studies were performed with a Hitachi F-7000 spectrometer (Tokyo, Japan); the excitation and emission slit widths were 5 nm; the photomultiplier (PMT) voltage was 400 V. In figures 4 and 5, the fluorescence images were captured using a Leica application suite advanced fluorescence (LAS-AF) instrument with a Leica TCS SP5. The light source was a white-light laser.
4.3. High-performance liquid chromatography separations
Several chromatographic systems were used for the analytical experiments and the purification steps: System A: RP-HPLC (Thermo Hypersil GOLD C18 column, 5 μm, 2.1 × 100 mm) with CH3CN and 0.1% aq. triethylammonium acetate (TEAA, 25 mM, pH 7.0) as eluents [0% CH3CN (2.5 min), followed by linear gradient from 0% to 80% (35 min) of CH3CN] at a flow rate of 0.25 mL min−1. Triple UV–vis detection was achieved at 230, 254, and 666 nm and with the 'Max Plot' mode (i.e. chromatogram at the absorbance maximum for each compound, 220–700 nm). System B: Semi-preparative RP-HPLC (Varian Kromasil C18 column, 10 μm, 21.2 × 250 mm) with CH3CN and aq. triethylammonium bicarbonate (aq. TEAB, 50 mM, pH 7.5) as eluents [0% CH3CN (5 min), followed by linear gradient from 0% to 100% (80 min) of CH3CN] at a flow rate of 20.0 mL min−1. Single visible detection was achieved at 570 nm. System C: System A with aq. TFA 0.1% as aq. mobile phase. System D: System B with aq. TFA 0.1% as aq. mobile phase.
Please note: SR101-NaphtNH2-Hyp-X (X = sulfobetaine and ammonium) derivatives were found to be soluble in D2O but low quality spectra were obtained (i.e. broad and poor-resolved peaks). Thus, the NMR spectra of such water-soluble derivatives were recorded in DMSO-d6.
4.4. Synthesis of SR101-NaphtNH2-Hyp-diMe
SR101-NaphtNH2 (TFA salt, 50 mg, 87 μmol, 1 equiv.) and TEA (12 μL, 38 μmol, 1 equiv.) were dissolved in dry CH3CN (1 mL) and the mixture was cooled to 0 °C and kept under an Ar atmosphere. Then, solid NOBF4 (15 mg, 130 μmol, 1.5 equiv.) was added and the reaction mixture was stirred at 0 °C for 15 min. Thereafter, a NaOAc buffer (0.1 M, pH 4.0, 0.5 mL) was added both to obtain complete solubilization of the diazonium salt (or N-nitroso intermediate) and to quench excess NOBF4. N,N-Dimethylaniline (11.6 mg, 95.7 μmol, 1.1 equiv.) was dissolved in CH3CN (0.1 mL) and this solution was added dropwise to the preformed diazonium salt (or N-nitroso) intermediate. The reaction mixture was stirred at rt for 30 min. The reaction was checked for completion by RP-HPLC (system A). Thereafter, the crude mixture was diluted with aq. TEAB (ca. 1.5 mL) and purified by semi-preparative RP-HPLC (system B). The product-containing fractions were lyophilized three times to give the TEA salt of SR101-NapthNH2-Hyp as a dark blue amorphous powder (36 mg, yield 51%). IR (neat): ν = 1644, 1613, 1557, 1487, 1396, 1354, 1320, 1234, 1033, 830, 693, 611 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 8.49 (d, 3J = 9.0 Hz, 1H), 8.31 (d, 4J = 1.6 Hz, 1H), 8.24 (s, 1H), 8.02 (d, 3J = 9.0 Hz, 1H), 7.94–7.81 (m, 2H), 7.78 (d, 3J = 9.0 Hz, 2H), 7.35 (d, 3J = 7.8 Hz, 1H), 7.05 (d, 3J = 8.9 Hz, 1H), 6.9–6.75 (m, 3H), 3.68 (m, 1H), 3.56 (m, 2H), 3.1 (q, 3J = 7.3 Hz, 6H, CH2-TEA), 3.08 (s, 6H), 2.60 (m, 2H), 2.11 (m, 2H), 1.83 (m, 2H), 1.16 (t, 3J = 7.3 Hz, 9H, CH3-TEA) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 157.0, 153.8, 153.1, 152.6, 152.0, 149.7, 149.3, 146.8, 142.7, 136.3, 129.1, 129.0, 128.1, 127.6, 126.0, 125.9, 125.4, 125.3, 125.2, 123.9, 123.4, 121.7, 119.6, 118.9, 118.9, 111.7, 104.8, 58.0, 51.3, 50.9, 45.8, 26.6, 19.6, 19.0, 18.7, 8.7 ppm; LRMS (ESI-): calcd for C37H31N7O7 [M—H]− 707.16, found: 707.27; UV–vis (PBS, pH 7.4, 25 °C): λmax = 578 nm (not obtained in a sufficient amount for a highly accurate determination of molar absorptivity); HPLC (system A): tR = 28.0 min, purity > 99%.
[M—H]− 707.16, found: 707.27; UV–vis (PBS, pH 7.4, 25 °C): λmax = 578 nm (not obtained in a sufficient amount for a highly accurate determination of molar absorptivity); HPLC (system A): tR = 28.0 min, purity > 99%.
4.5. Synthesis of SR101-NaphtNH2-Hyp-diN3
The same procedure described above was used. N,N-di(2-azidoethyl)aniline 3 (22.1 mg, 95.7 μmol, 1.1 equiv.) was used for this azo coupling. The product-containing fractions were lyophilized three times to give the TEA salt of SR101-NaphtNH2-Hyp-diN3 as a dark blue amorphous powder. (35 mg, yield 44%). IR (neat): ν = 2101, 1644, 1611, 1549, 1425, 1486, 1394, 1340, 1321, 1239, 1197, 1171, 1032, 831, 751, 691, 610 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 8.59 (d, 3J = 8.5 Hz, 1H), 8.35 (bs, 1H), 8.31 (d, 4J = 1.6 Hz, 1H), 8.11 (d, 3J = 8.9 Hz, 1H), 7.95–7.8 (m, 4H), 7.35 (d, 3J = 7.8 Hz, 1H), 7.05 (m, 4H), 6.84 (s, 1H), 3.75 (m, 6H), 3.61 (m, 6H), 3.14 (bs, 2H), 3.06 (q, 3J = 7.3 Hz, 6 H, CH2-TEA), 2.62 (m, 2H), 2.15 (bs, 2H), 1.84 (bs, 2H), 1.16 (t, 3J = 7.3 Hz, 9H, CH3-TEA) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 162.9, 157.0, 153.9, 152.5, 152.1, 150.7, 149.6, 149.3, 146.8, 143.4, 136.2, 129.0, 128.9, 128.1, 127.8, 126.0, 125.8, 125.5, 125.2, 123.9, 121.9, 119.6, 119.1, 112.2, 111.2, 104.9, 51.3, 50.9, 49.3, 48.2, 48.0, 45.7, 26.6, 19.6, 19.1, 18.8, 8.6 ppm; LRMS (ESI-): calcd for C39H33N10O7 [M—H]− 817.20, found: 817.13. UV–vis (PBS, pH 7.4, 25 °C): λmax = 573 nm (not obtained in a sufficient amount for a highly accurate determination of molar absorptivity); HPLC (system A): tR = 29.3 min, purity = 85%.
[M—H]− 817.20, found: 817.13. UV–vis (PBS, pH 7.4, 25 °C): λmax = 573 nm (not obtained in a sufficient amount for a highly accurate determination of molar absorptivity); HPLC (system A): tR = 29.3 min, purity = 85%.
4.6. Synthesis of SR101-NaphtNH2-Hyp-sulfobetaine
SR101-NaphtNH2-Hyp-diN3 (10 mg, 12 μmol, 1 equiv.) and terminal alkyne 4 (6.4 mg, 26 μmol, 2.2 equiv.) were dissolved in 500 μL of a (1: 1, v/v) mixture of DMF/H2O. Sodium ascorbate (1.2 mg, 6 μmol, 0.5 equiv.) and CuSO4. 5 H2O (0.75 mg, 3 μmol, 0.25 equiv.) were sequentially added and the resulting reaction mixture was stirred at rt under an Ar atmosphere for 2 h. The reaction was checked for completion by RP-HPLC (system A). Thereafter, the crude mixture was diluted with aq. TEAB (ca. 1.5 mL) and purified by semi-preparative RP-HPLC (system B) to give after lyophilization the TEA salt of SR101-NaphtNH2-Hyp-sulfobetaine, as a dark blue amorphous powder (11 mg, yield 65%). IR (neat): ν = 3375, 2949, 1645, 1599, 1556, 1483, 1422, 1339, 1319, 1144, 1027, 874, 827, 690, 606 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 8.81 (s, 2H), 8.62 (d, 3J = 9.0 Hz, 1H), 8.38 (s, 1H), 8.31 (s, 1H), 8.14 (d, 3J = 9.1 Hz, 1H), 7.85 (d, 3J = 9.1 Hz, 1H), 7.82 (d, 3J = 8.9 Hz, 1H), 7.75 (m, 2H), 7.32 (d, 3J = 7.7 Hz, 1H), 7.07 (d, 3J = 7.7 Hz, 1H), 6.93 (d, 3J = 9.0 Hz, 1H), 6.87 (s, 1H), 4.65 (bs, 8H), 3.9 (bs, 4H), 3.72 (bs, 2H), 3.63 (bs, 2H), 3.25 (m, 8H), 3.06 (q, 3J = 7.3 Hz, 6H, CH2-TEA), 2.70 (m, 6H), 2.15 (m, 6H), 1.83 (m, 10H), 1.53 (m, 4H), 1.16 (t, 3J = 7.3 Hz, 9H, CH3-TEA) ppm; A good quality 13C spectrum could not be obtained despite the extended acquisition time (more than 80 000 scans) on a 500 MHz spectrometer (equipped with a 5 mm 'BBFO' broad band ATMA gradient z probe four times more sensitive than a standard 300 MHz spectrometer). This is due to the moderate solubility of SR101-NaphtNH2-Hyp-sulfobetaine (even under its TEA salt form) in DMSO-d6 and molecular motions around the azo bond. LRMS (ESI-): calcd for C61H71N12O13 [M—H]− 1308.42, found: 1308.40; UV–vis (PBS, pH 7.4, 25 °C): λmax = 570 nm (not obtained in a sufficient amount for a highly accurate determination of molar absorptivity); HPLC (system A): tR = 23.5 min, purity = 98%. It is possible to make desalting over Dowex H+ resin to obtain the free sulfonic acid form (8.2 mg, yield 52%).
[M—H]− 1308.42, found: 1308.40; UV–vis (PBS, pH 7.4, 25 °C): λmax = 570 nm (not obtained in a sufficient amount for a highly accurate determination of molar absorptivity); HPLC (system A): tR = 23.5 min, purity = 98%. It is possible to make desalting over Dowex H+ resin to obtain the free sulfonic acid form (8.2 mg, yield 52%).
4.7. Synthesis of SR101-NaphtNH2-Hyp-ammonium
SR101-NaphtNH2-diN3 (10 mg, 12 μmol, 1 equiv.) and terminal alkyne 5 (5.3 mg, 26 μmol, 2.2 equiv.) were dissolved in 500 μL of a (1: 1, v/v) mixture of DMF/H2O. Sodium ascorbate (1.2 mg, 6 μmol, 0.5 equiv.) and CuSO4. 5 H2O (0.75 mg, 3 μmol, 0.25 equiv.) were sequentially added and the resulting reaction mixture was stirred at rt under an Ar atmosphere for 2 h. The reaction was checked for completion by RP-HPLC (system C). Thereafter, the crude mixture was diluted with aq. 0.1% TFA (ca. 1.5 mL) and purified by semi-preparative RP-HPLC (system D) to give after lyophilization the TFA salt of SR101-NaphtNH2-Hyp-ammonium as a dark blue amorphous powder (9.1 mg, yield 71%). IR (neat): ν = 1686, 1646, 1595, 1513, 1489, 1460, 1442, 1383, 1363, 1319, 1198, 1161, 1128, 1076, 1055, 1032, 1013,829, 800, 784, 759, 719, 686, 672, 610 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 8.47 (s, 2H), 8.35 (m, 2H), 8.22 (bs, 1H), 7.92 (m, 2H), 7.80 (d, 3J = 9.1 Hz, 1H), 7.64 (m, 2H), 7.38 (d, 3J = 9.0 Hz, 1H), 7.03 (d, 3J = 7.7 Hz, 1H), 6.79 (m, 3H), 4.65 (m, 8H), 3.90 (bs, 4H), 3.68 (bs, 2H), 3.59 (bs, 2H), 3.41 (m, 6 H), 3.28 (m, 6H), 2.98 (bs, 2H), 2.87 (s, 6H), 2.04 (m, 12H), 1.85 (m, 2H) ppm; a good quality 13C spectrum could not be obtained despite the extended acquisition time (more than 80 000 scans) on a 500 MHz spectrometer (equipped with a 5 mm 'BBFO' broad band ATMA gradient z probe four times more sensitive than a standard 300 MHz spectrometer). This is due to the moderate solubility of SR101-NaphtNH2-Hyp-ammonium in DMSO-d6 and molecular motions around the azo bond. 13C NMR (126 MHz, DMSO-d6): δ = 156.8, 154.0, 152.5, 152.1, 150.6, 149.7, 149.3, 146.8, 143.4, 136.1, 132.4, 130.2, 129.1, 128.6, 128.2, 127.9, 127.5, 126.0, 125.8, 125.3, 123.9, 123.8, 122.0, 119.7, 119.2, 119.1, 111.6, 104.9, 62.8, 56.3, 51.4, 51.0, 48.8, 48.0, 47.1, 30.7, 26.7, 21.1, 19.7, 19.1, 18.8 ppm; LRMS (ESI+): calcd for C55H61N12O7 [M]+· 1065.42, found: 1065.73; UV–vis (PBS, pH 7.4, 25 °C): λmax = 570 nm (not obtained in a sufficient amount for a highly accurate determination of molar absorptivity); HPLC (System B): tR = 23.8 min, purity > 99%.
[M]+· 1065.42, found: 1065.73; UV–vis (PBS, pH 7.4, 25 °C): λmax = 570 nm (not obtained in a sufficient amount for a highly accurate determination of molar absorptivity); HPLC (System B): tR = 23.8 min, purity > 99%.
4.8. Preparation of rat liver microsomes and in vitro assay
The rats (Wistar, ♂ 6–7 weeks) were purchased from CLEA Japan. The rats received an intraperitoneal injection of 60 mg kg−1 sodium phenobarbital once daily for three days, then they were fasted overnight and sacrificed by exsanguination from the abdominal aorta. The liver containing 0.15 M KCl (pH 7.4) was homogenized in 3 volumes of the same buffer. The microsomes were prepared according to the method of Omura and Sato [30]. The hypoxic condition in vitro (enzyme assay in cuvette) was prepared by bubbling argon gas into the reaction solution (0.1 M potassium phosphate buffer, pH 7.4) for 30 min. The rat liver microsomes (226 μg/3 mL) were pre-incubated at 37 °C for 5 min and then 1 μM azo-based probes SR101-NaphtNH2-X containing 0.1% DMSO as a cosolvent was added. As a cofactor for reductases, 50 μM NADPH was added at 3 min.
4.9. Cell lines and culture conditions, hypoxic conditions for living cell fluorescence imaging and fluorescence confocal microscopy
4.9.1. Cell lines and culture conditions
Human lung carcinoma cell line A549 was purchased from RIKEN Bioresource Center cell bank (Tsukuba, Japan). A549 cells were cultured in DMEM (Dulbecco's modified Eagle's medium) (Invitrogen) containing 10% fetal bovine serum (Invitrogen) and 1% penicillin streptomycin (Invitrogen). The cells were maintained at 37 °C under 5% CO2 in air as a standard condition.
4.9.2. Hypoxic conditions for live cell fluorescence imaging
An O2 concentration of 0.1% was generated with an Anaero Pack® (Mitsubishi Gas Chemical Company, Inc.) and a 2.5 L rectangular jar (Mitsubishi Gas Chemical Company, Inc.). The O2 concentration in the range of 1–20% was controlled with a multi-gas incubator (Sanyo) by means of N2 substitution.
4.9.3. Fluorescence confocal microscopy
The details of the confocal fluorescence imaging of A549 cells under hypoxia were reported previously [15]. 3 × 104 A549 cells were seeded on 8-chamber plates (NUNCTM) and cultured for one day before assay. The cells were washed with PBS once, and then incubated in 200 μL DMEM containing a 1 μM azo-based probe SR101-NaphtNH2-X and 0.1% DMSO as a cosolvent at various oxygen levels. Fluorescence confocal microscopic images were acquired by using a Leica Application Suite Advanced Fluorescence (LAS-AF) instrument with a TCS SP5 and a 40 × or 10 × objective lens. The light source was a white-light laser.
Acknowledgments
Funding: The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors acknowledge financial support from INSA Rouen, Rouen University, CNRS, Labex SynOrg (ANR-11-LABX-0029) and région Haute-Normandie (CRUNCh network), and JSPS KAKENHI Grant Number 26104509 to K H Financial support from FEDER (TRIPODE, no. 33883) for the PhD grant of A C is also gratefully acknowledged. A R thanks the Institut Universitaire de France (IUF) and the Burgundy region ('FABER' programme, PARI Action 6, SSTIC 6 'Imagerie, instrumentation, chimie et applications biomédicales') for their financial support, and the 'Plateforme d'Analyse Chimique et de Synthèse Moléculaire de l'Université de Bourgogne (PACSMUB, www.wpcm.fr)' for the access to Bruker Avance III 500 NMR and Bruker Alpha FT-IR spectrometer. We also thank Patricia Martel (University of Rouen, laboratory COBRA, UMR CNRS 6014) for the recording of the IR spectra.
Competing interests
The authors have declared that no competing interests exist.
Footnotes
- 5
The term 'latent fluorophore' was introduced by Raines et al see [11].